CHAPTER 20
Fetal Cardiomyopathies
Marisa R. Lydia and Julia A. Drose
Definition
A cardiomyopathy is a disorder of heart muscle that may be a primary disorder or may be associated with structural anomalies or pericardial disease. Cardiomyopathies account for approximately 2% of congenital heart disease in live-born infants.1,2 The in utero occurrence is reported to be between 8% and 11%.3 There are three different presentations in the fetus. The congestive, or dilated, form is most common and presents with dilated, poorly contracting chambers, atrioventricular regurgitation, and, often, associated hydrops fetalis (pleural and pericardial effusions, ascites, and skin thickening). The second most common type of cardiomyopathy is the hypertrophic form. Hypertrophic cardiomyopathies are recognized by markedly thickened ventricular walls and septum. The third form, a restrictive cardiomyopathy, is unusual in the fetus, manifesting primarily as endocardial fibroelastosis.4,5
Physiology
Fetal cardiomyopathies are caused by a broad spectrum of underlying disorders (Table 20–1). There is not always correlation between presentation (i.e., dilated versus hypertrophic) and cause; however, it is helpful to use basic physiological principles to understand, identify, and perhaps treat the underlying disorder.
Dilated Cardiomyopathies
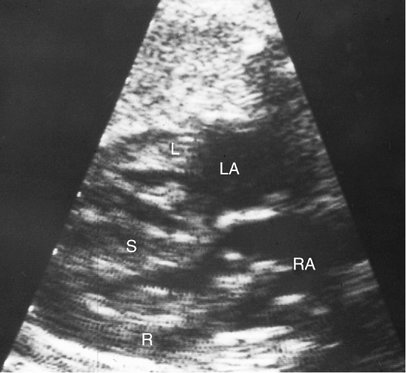
Severe fetal anemias result in high-output failure when the fetal heart increases cardiac output by increasing heart rate or stroke volume, or both, to meet peripheral oxygen requirements. As a result, cardiac work and oxygen demand increase and eventually exceed myocardial oxygen supply. Myocardial ischemia leads to poor cardiac function and contractility with chamber dilation (Fig. 20–1).
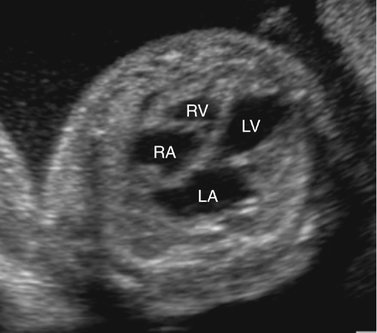
Anemia-associated cardiomyopathies include immune hydrops fetalis, homozygous alpha-thalassemia, glucose-6-phosphate-dehydrogenase deficiency, and hemophilia A.6 Immune hydrops fetalis occurs when maternal serum immunoglobulin (Ig) G antibodies are formed against antigens on fetal red blood cells. Maternal serum antibodies are formed from the mixing of maternal and fetal blood during parturition of a prior pregnancy, placental abruption, amniocentesis, spontaneous or therapeutic abortion, blood transfusion, or placental intervillous hemorrhage. Maternal IgG crosses the placenta, enters the fetal circulation, and binds to fetal red blood cell antigens. Fetal hemolysis and hemolytic anemia may then occur.
A nonimmune mediated anemia, homozygous alpha-thalassemia, accounts for approximately 10% of cases of nonimmune hydrops fetalis in North America.6 In this condition, fetal red blood cells contain Bart hemoglobin. This hemoglobin irreversibly binds to oxygen, resulting in the inability of red blood cells to deliver oxygen to fetal tissues. Homozygous alpha-thalassemia is uniformly fatal to the fetus. If the pregnancy is allowed to progress, there is a significant risk of preeclampsia and maternal microcytic anemia.
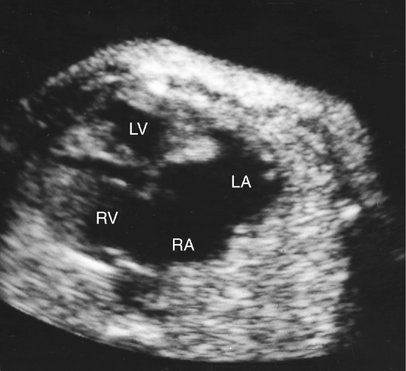
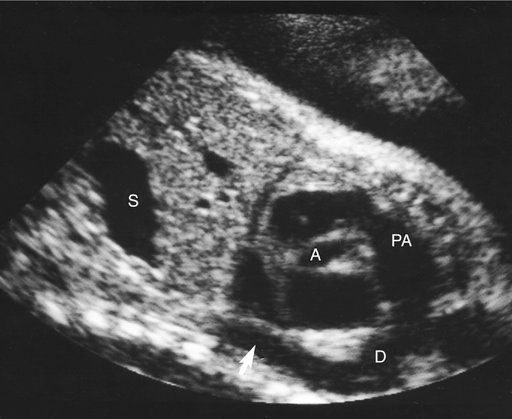
Hemangioendotheliomas of the liver, sacrococcygeal teratomas, intracranial teratomas, vein of Galen arteriovenous malformations, and other congenital shunts may present with dilated cardiomyopathy caused by high-output failure from volume overload (Figs. 20–2 and 20–3).6–11
Direct myocardial damage, another cause of dilated cardiomyopathy, includes fetal infection, tachycardia-induced cardiomyopathies, and fetal anoxia.12
Direct myocardial damage with resultant heart failure may occur with fetal infection (Fig. 20–4).13,14 Coxsackie virus; toxoplasmosis, rubella, cytomegalovirus, and herpes simplex (TORCH) agents; and parvovirus are most often responsible. Maternal penicillin therapy for syphilis may reverse syphilitic fetal myocarditis, congestive heart failure, and hydrops fetalis.14 Although identification of viral agents may be particularly difficult, cultures and serological tests, as well as associated sonographic findings (i.e., intracranial or hepatic calcifications, hepatosplenomegaly, echogenic bowel), may confirm suspected cases.
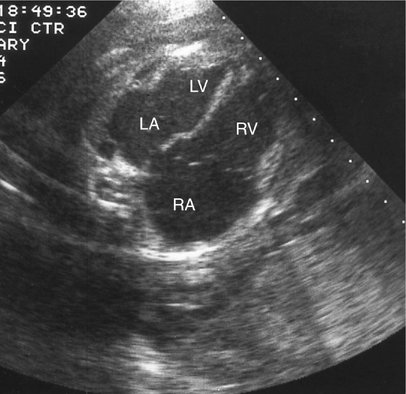
Myopathy induced by arrhythmia (tachyarrhythmia or bradyarrhythmia) is the most common cause of nonimmune hydrops fetalis in North America.15 Fetal supraventricular tachycardia is the most common sustained fetal arrhythmia. It may present with congestive cardiomyopathy when the rapid heart rate results in such shortened diastole that myocardial perfusion (the majority of which occurs in diastole) is markedly diminished (Fig. 20–5). Myocardial ischemia and direct myocardial cell damage may ensue.9 Fetal supraventricular tachycardia is one of the few potentially treatable causes of congestive heart failure and therefore is important to recognize to initiate appropriate therapy.16,17
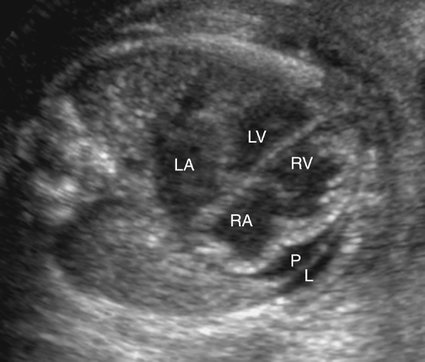
Fetal asphyxia can result in direct myocardial damage and cardiac decompensation.18,19 The fetus responds to hypoxia by vasoconstriction, bradycardia, and hypertension. Hypotension, myocardial ischemia, and myocardial cell injury occur with worsening asphyxia, resulting in subsequent cardiac decompensation.
Hypertrophic Cardiomyopathies
Hypertrophic cardiomyopathies present with thickened ventricular walls from increased myocardial cell size, resulting in decreased ventricular compliance (Fig. 20–6). This may lead to poor diastolic filling of the heart (diastolic dysfunction), which, in turn, results in diminished cardiac output.
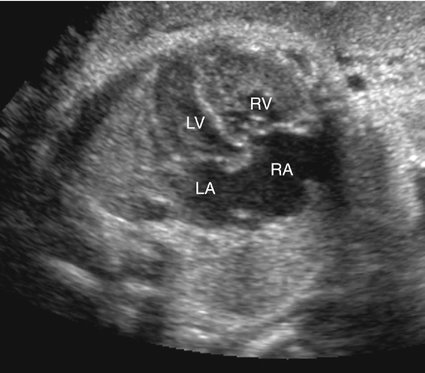
Biventricular hypertrophy occurs in most recipient twins in twin-twin transfusion syndrome (TTTS) (Fig. 20–7).20,21 In this syndrome, monochorionic twins share (usually at the level of the cotyledons) a placental arteriovenous circulation. Blood is transferred through the interconnected placental circulation from a donor twin on the arterial side to a recipient twin on the venous side. This results in an oligohydramniotic, anemic donor twin and a plethoric, polyhydramniotic and often hydropic recipient twin. Although the cardiomegaly found in the recipient twin has long been thought to result from volume overload, no significant difference in cardiac output, when adjusted to fetal weight, is found between donor and recipient.20 The fact that the cardiomegaly present in the recipient twin exhibits myocardial hypertrophy rather than ventricular dilation implies that the pathogenesis is more complex. Therapies used to treat TTTS in utero, such as amnio reduction or anastomosis ablation, do not seem to improve cardiac function. In fact, the hypertrophic cardiomyopathy associated with TTTS often persists and worsens as gestation progresses.22 Donor twins are affected with cardiovascular disease much less frequently.20
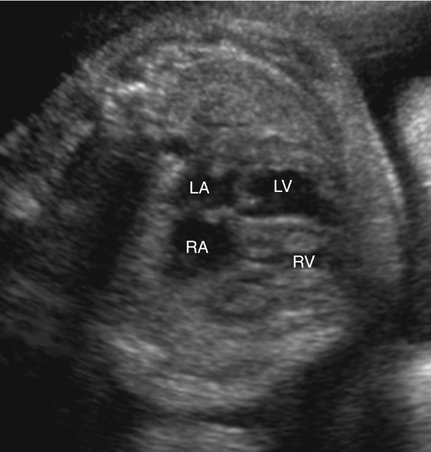
Fetal hypertrophic cardiomyopathy can also occur as a result of maternal insulin-dependent diabetes. The incidence of hypertrophic cardiomyopathy in infants of diabetic mothers has been reported to be from 33% to 84%.23–27 In this condition, the ventricular septum can be disproportionately thickened in relation to the ventricular free walls (Fig. 20–8), similar to the asymmetrical septal hypertrophy (ASH) (unrelated to diabetes) seen in children and adults. Concentric hypertrophy may also occur. Infants of diabetic mothers may also have fetal hyperinsulinemia, macrosomia, and organomegaly. There is conflicting opinion in the literature regarding the pathophysiologic mechanisms of these changes.24,27–28 One theory is that maternal hyperglycemia induces production of fetal insulin. The anabolic effect of endogenous fetal insulin subsequently causes macrosomia and organomegaly, including ASH.
Generally, ASH is progressive throughout gestation. Fetuses may therefore appear normal in second-trimester examinations and subsequently exhibit sonographic findings of diabetic hypertrophic cardiomyopathy on follow-up examination. ASH is usually asymptomatic in the fetus and neonate, with spontaneous postnatal resolution.29,30 Rarely, hypertrophic cardiomyopathy is a primary disorder of hypertrophic myocardium, as is seen in Noonan syndrome and glycogen storage disease.3,6,31
Restrictive Cardiomyopathies
Restrictive cardiomyopathies decrease cardiac output because of impaired diastolic filling.32 In the fetus, this is primarily due to abnormal fibroelastic proliferation within the endocardium (endocardial fibroelastosis). This abnormal tissue decreases endocardial compliance, with resultant diastolic dysfunction and decreased cardiac output. Endocardial fibroelastosis results from pathological deposition of collagen and elastin within the endocardium.33 It accounts for approximately 1% to 4% of cases of congenital heart disease and occurs in 1 of every 5000 to 6000 births.34 It is divided into primary and secondary forms.
Primary endocardial fibroelastosis occurs when no associated structural cardiac anomaly is found. It may be a result of fetal infection such as Coxsackie virus or of an autoimmune process such as lupus erythematosus. In this setting, transplacental passage of maternal anti-Ro and anti-La antibodies induce inflammation and fibrosis of the myocardium.33,35,36
Secondary endocardial fibroelastosis is usually associated with obstructive lesions, predominantly left sided, such as aortic stenosis or atresia.36 In this setting, obstruction to blood flow causes the fibrin build up, which is then deposited in the endocardium.33,34 The sonographic appearance is classically ventricular enlargement and a strongly echogenic endocardium (Fig. 20–9). Contracted forms have been reported without ventricular enlargement, and there is at least one report of a dilated endocardial fibroelastosis progressing to a contracted form. Therapy includes digoxin and diuretics, but the prognosis is poor. Most fetuses die in utero or in the early neonatal period.37–41


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


