Outline
Embryology, 504
Normal Fetal Urinary Tract, 504
Obstructive Urinary Tract Abnormalities, 504
Transient Dilation, 506
Ureteropelvic Junction Obstruction, 506
Ureterovesical Junction Obstruction, 509
Duplex Collecting System/Ureterocele, 509
Vesicoureteral Reflux, 511
Lower Urinary Tract Obstruction, 512
Urethral Atresia, 515
Congenital Megalourethra, 515
Cloacal Malformation, 515
Prune-Belly Syndrome, 515
Megacystis-Microcolon–Intestinal Hypoperistalsis Syndrome, 515
Management of Fetal Renal Pelvic Dilatation After Birth, 516
Nonobstructive Fetal Urinary Tract Anomalies, 517
Nonobstructive Megaureter, 517
Empty Renal Fossa, 518
Unilateral Renal Agenesis, 519
Aberrant Cephalad Migration, 520
Renal Fusion Abnormalities, 520
Dysplastic Kidneys, 521
Miscellaneous Cystic Renal Disorders, 523
Unclassified Renal Cystic Diseases, 523
Multicystic Dysplastic Kidney, 523
Renal Tumors, 525
Nonobstructive Fetal Bladder Abnormalities, 526
Bilateral Single Ectopic Ureters, 528
Fetal Adrenal Glands, 529
Fetal Genital Anomalies, 531
Disorders of Sexual Development, 532
Fetal Ovarian Cysts, 532
Hydrocolpos/Hydrometrocolpos, 534
Summary of Key Points
- •
Normal renal function is fully established by the 9th week of gestation.
- •
It is preferable to describe the findings in the fetal renal pelvis, calyces, ureter, and bladder in cases of urinary tract dilation, rather than using nonspecific terms such as pyelectasis, caliectasis, and hydronephrosis.
- •
Renal cystic anomalies may be a manifestation of underlying genetic disorders.
- •
Ultrasound findings in cases of lower urinary tract obstruction (LUTO) are more predictive of poor renal function than serial biochemical analyses of fetal urine.
- •
Although inability to visualize a kidney in its normal location is typically associated with either renal agenesis or ectopia, an abnormal or nonvisualized normal kidney remains a possibility.
- •
In sonographic terms, dysplasia is nonspecific and refers to a variety of findings.
- •
Over 50% of all congenital abdominal masses found in the neonate originate in the kidney with the most common fetal renal tumor being mesoblastic nephroma.
- •
Prominent adrenal glands suggest either renal agenesis, congenital adrenal hyperplasia (CAH), or the presence of a de novo neoplastic process.
- •
Disorders of sexual differentiation are heterogeneous and may be secondary to hormonal defects or unidentified genetic diseases.
- •
Fetal ovarian cysts have an excellent prognosis as the majority resolve in the postnatal period spontaneously.
Congenital abnormalities of the kidneys and urinary tract (CAKUT) affect 1 in 500 live births and constitute about 20% of all major fetal anomalies, and autopsy evidence suggests an even higher prevalence. Although antenatal diagnosis of urinary tract malformations appears to be improving secondary to improved resolution of ultrasound equipment, there are relatively high false positive rates. This is compounded by the lack of a consensus on terminology and diagnostic criteria for cases of antenatal urinary tract dilatation.
Embryology
The urogenital system consists of two structurally and functionally different components—the urinary system and the genital system. Both develop from a common ridge of mesodermal tissue, the intermediate mesoderm, along the posterior wall of the abdominal cavity.
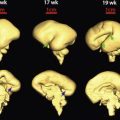
Human embryos develop three sets of excretory organs or kidney systems during intrauterine life. The embryonic kidneys, which form in a cranial to caudal sequence, in order of appearance are the pronephros, the mesonephros, and the metanephros. The pronephros and the mesonephros both regress in utero, and the metanephros ultimately becomes the permanent kidneys ( Fig. 15-1A and B ). The mesonephros serves as an excretory organ for the embryo whereas the definitive kidney begins its development, then regresses by the 4th month. Some elements of the mesonephros are retained in the mature urogenital system as part of the reproductive tract.

The metanephros forms in the sacral region as a pair of structures called the metanephric diverticulum or ureteric bud. It emanates from the distal portion of the mesonephric duct and comes in contact with and penetrates the metanephric mesenchymal blastema (metanephric mesoderm) at approximately the 28th day.
The ureteric bud and metanephric mesoderm exert reciprocal inductive effects toward each other, and the proper differentiation of these structures depends on these inductive signals. The metanephric mesoderm induces the ureteric bud to branch, and the ureteric bud induces the metanephric mesoderm to condense and undergo mesenchymal-epithelial conversion. The nephron, which consists of the glomerulus, proximal tubule, loop of Henle, and distal tubule, is thought to derive from the metanephric mesoderm, whereas the collecting system, consisting of collecting ducts, calyces, pelvis, and ureter, is formed from the ureteric bud.
The division of the ureteric bud results in the eventual pelvicalyceal patterns and their corresponding renal lobules. The first few divisions of the ureteric bud give rise to the renal pelvis, major and minor calyces, and collecting tubules. Thereafter, the first generations of collecting ducts are formed. When the ureteric bud first invades the metanephric mesoderm, its expanded end forms an ampulla that will eventually give rise to the renal pelvis. By the 6th week, the ureteric bud has bifurcated at least four times, yielding 16 branches. These branches then coalesce to form two to four major calyces extending from the renal pelvis. By the 7th week, the next four generations of branches also fuse, forming the minor calyces. By the 32nd week, approximately 11 additional generations of bifurcation have resulted in approximately 1 to 3 million branches, which will become the collecting duct tubules. In humans, although renal maturation continues to take place postnatally, nephrogenesis is completed before birth. The kidney moves cephalad from its pelvic position to the lumbar region by the 6th to 9th week of gestation.
Initially, the excretory ducts of both systems enter a common cavity, the cloaca. The urorectal septum develops during the 4th to 6th weeks and separates the cloaca into an anterior urogenital sinus and the rectum posteriorly. The urogenital sinus gives rise to the bladder superiorly and the membranous urethra inferiorly. Initially the bladder is continuous with the allantois, but when the lumen of the allantois is obliterated, a thick fibrous cord, the urachus, remains and connects the apex of the bladder with the umbilicus. The genital ridges can be seen medial to the mesonephros and these form the gonads, which differentiate into male and female testis and ovaries, respectively, by the 8th week.
Normal Fetal Urinary Tract
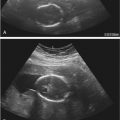
With the use of transvaginal transducers, the fetal kidneys can be demonstrated at approximately 11 weeks, and they can be visualized by 12 weeks using transabdominal probes ( Fig. 15-2 ). During the first trimester, the kidneys appear as hyperechoic oval structures at both sides of the spine with echogenicity comparable to that of the liver or spleen. The bladder can be seen in most fetuses from the 9th week of gestation ( Fig. 15-3 ). The echogenicity of the fetal kidneys progressively decreases, and by the third trimester, the cortical echogenicity should be less than that of the liver or spleen. Corticomedullary (CMD) differentiation begins at about 14 to 15 weeks and should always be demonstrated in fetuses older than 18 weeks ( Fig. 15-4B ). Growth and appropriate size of the fetal kidneys can be evaluated by measuring renal length and comparing it to normal charts. The normal kidney grows at an average rate of 1.1 mm per gestational week. During the second and third trimesters, the kidneys are easily identified by imaging the dorsolumbar spine and scanning on either side in parasagittal and transverse axial sections ( Fig. 15-4A ). Urine distending the renal pelvis may help in their identification. Under normal conditions, the fetal ureters are not visible.



Production of urine by the kidneys begins during the 9th week of embryonic life. At this stage, the urine in the bladder allows visualization as a fluid-filled structure within the fetal pelvis ( Fig. 15-5 ).

During the second and third trimester, the bladder will empty and refill continuously every 25 to 30 minutes. This cycle can be monitored during the sonographic examination. Toward the end of the pregnancy, this cycle decreases, especially in female fetuses, possibly from hormonal influence on the fetal bladder neck. The position of the fetal bladder is easily identified, because it lies between the umbilical arteries within the fetal pelvis. These arteries are readily seen with the use of color Doppler imaging. The normal thickness of the bladder wall is about 2 mm, and the ideal position to measure this is at the level of the umbilical arteries.
In addition to visualization of the bladder and normal kidneys, the assessment of the urinary tract (UT) should include an evaluation of the amniotic fluid volume. After 14 weeks, two thirds of the normal amniotic fluid is produced by fetal urination and one third comes from pulmonary fluid. A normal volume of amniotic fluid is mandatory for proper development of the fetal lung.
Obstructive Urinary Tract Abnormalities
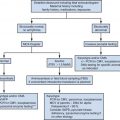
Urinary tract dilation is one of the most common sonographic prenatal diagnoses, affecting 1% to 5% of all pregnancies. The condition is most commonly diagnosed by measuring the anteroposterior (AP) diameter of the renal pelvis on a transverse scan of the fetal abdomen ( Figs. 15-6 and 15-7 ). The exact incidence is confounded by the different terms used to describe this condition in the literature, including hydronephrosis, pyelectasis, pelviectasis, and pelvicaliectasis. In an attempt to limit the use of these confusing terms, this chapter will use the term urinary tract dilation and be more specific when the underlying renal abnormality is known, as recently suggested in a Consensus Statement endorsed by the major fetal and postnatal renal imaging societies. The proposed classification system suggests describing findings on renal ultrasound examination as shown in Table 15-1 and Figure 15-8 and classifying normal renal findings as in Table 15-2 . The panel went on to propose stratifying prenatal risks and management schemes based on findings as shown in Figure 15-9 . Examples of sonographic features of common findings associated with urinary tract dilation are shown in Figures 15-8 and 15-10A and B .


| Ultrasound Parameters | Measurement/Findings | Note |
|---|---|---|
| Anteroposterior renal pelvic diameter (APRPD) | AP in mm | Measured on transverse image at the maximal diameter of intrarenal pelvis ( Fig. 15-7 ) |
| Calyceal dilation: central (major calyces) | Yes/no | |
| Calyceal dilation: peripheral (minor calyces) | Yes/no | |
| Parenchymal thickness | Normal/abnormal | Subjective assessment |
| Parenchymal appearance | Normal/abnormal | Evaluate echogenicity, corticomedullary differentiation, and for cortical cysts |
| Ureter | Normal/abnormal | Dilation of ureter is considered abnormal prenatally, although transient visualization of the ureter is considered normal postnatally |
| Bladder | Normal/abnormal | Evaluate wall thickness for the presence of ureterocele and for a dilated posterior urethra |

| Ultrasound Findings | TIME AT PRESENTATION | ||
|---|---|---|---|
| 16 to 27.9 Weeks | ≥28 Weeks | Postnatal (>48 Hours) | |
| Anteroposterior renal pelvic diameter | <4 mm | <7 mm | <10 mm |
| Calyceal dilation | |||
| Central | No | No | No |
| Peripheral | No | No | No |
| Parenchymal thickness | Normal | Normal | Normal |
| Parenchymal appearance | Normal | Normal | Normal |
| Ureter(s) | Normal | Normal | Normal |
| Bladder | Normal | Normal | Normal |
| Unexplained oligohydramnios | No | No | NA |


In the low-risk group, other than one prenatal follow-up sonogram, no postnatal follow-up is recommended. This approach is in contrast to high-risk groups, which require closer and more frequent evaluation. The proposed classification system builds on that previously proposed by the Society for Fetal Urology ( Fig. 15-11A and B ), with the advantage of being endorsed by other professional organizations, including the Society for Maternal-Fetal Medicine, the American Institute for Ultrasound in Medicine, and the American College of Radiologists. Prospective studies are needed to evaluate the effectiveness of the classification system in streamlining management of these abnormalities. The common causes, and their incidence, of urinary tract dilation are shown in Table 15-3 .

| Cause | Incidence |
|---|---|
| Transient dilation | 41-88% |
| Ureteropelvic junction (UPJ) obstruction | 10-30% |
| Vesicoureteral reflux (VUR) | 10-20% |
| Ureterovesical junction (UVJ) obstruction | 5-10% |
| Duplex collecting system/ureterocele | 5-7% |
| Multicystic dysplastic kidney | 4-6% |
| Lower urinary tract obstruction (LUTO) | 1-2% |
Transient Dilation
The majority of fetuses with an antenatal diagnosis of renal dilation have normal renal findings postnatally. Transient dilation is thought to result from narrowing or natural folds within the urinary tract during the early developmental phase, which eventually resolve. Because this is a retrospective diagnosis, it is difficult to differentiate from other causes of urinary tract dilation. In general, the fetuses with transient dilation tend to have AP diameters of less than 6 mm in the second trimester and less than 8 mm in the third trimester.
Ureteropelvic Junction Obstruction
The hallmark of ureteropelvic junction (UPJ) obstruction is the finding of a dilated renal pelvis and calyces without dilation of the ureters. This conditions affects 1 in 2000 births and is responsible for 10% to 30% of all cases of antenatal renal dilation. The cause of UPJ obstruction remains unknown, but it is observed more commonly in male fetuses.
The threshold measurement for diagnosis on axial views of the kidney is 7 mm for mild dilation (see Fig. 15-6 ), between 7 and 15 mm for moderate dilation, and greater than 15 mm for marked dilation by 32 weeks’ gestation. The more dilated the system, the more likely it is that renal function will be compromised after birth. In addition to measurement of the renal pelves, it is important to determine and to document whether the central or peripheral calyces are dilated. Evaluation of the kidneys should also include assessment of the appearance of the cortices; thinned, hyperechogenic renal cortices with cysts often represent obstructive dysplasia and impaired function ( Fig. 15-12 ). Although there is inconsistent correlation between the antenatal renal appearance and postnatal function, the presence of cysts commonly indicates the presence of dysplasia and compromised renal function.

The differential diagnosis of UPJ obstruction includes reflux, transient dilation, multicystic dysplastic kidney (MCDK), and ureterovesical junction (UVJ) obstruction. Less common differential diagnoses of UPJ obstruction are megacalycosis due to medullary hypoplasia in which the calyces are more dilated than the renal pelvis; infundibular stenosis with no medullary hypoplasia, but calyceal dilation still present; and isolated cysts within the kidney.
Associated renal or extrarenal anomalies are present in 12% to 25% of cases of UPJ obstruction. Once diagnosed in the second trimester, follow-up at or after 32 weeks is recommended to exclude cases with transient dilatation that may have resolved by this time (<7 mm dilated). Conflicting reports have been published regarding the association of isolated mild renal pelvic dilation and aneuploidy, but the overall evidence suggests that although this association is weak, it is significant and should be discussed with patients. Patients should be informed of the overall good prognosis for fetuses with UPJ obstruction. Of those cases that are persistent in the postnatal period, 19% to 25% may require some form of surgical intervention.
Ureterovesical Junction Obstruction
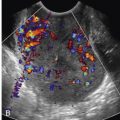
UVJ obstruction is responsible for 5% to 10% of all cases of urinary tract dilation. In about 25% of cases, the contralateral kidney is also affected. The diagnosis of UVJ obstruction is based on the demonstration of a dilated ureter in the presence of a dilated renal pelvis and a normal bladder. Peristaltic waves can often be seen, and they modify the caliber of the ureter ( Fig. 15-13 ). The dilatation may increase in utero, but it usually decreases after birth.

The underlying cause of UVJ obstruction appears to be a localized dysfunction in a region of the lower ureter. In most instances, it is not possible to differentiate between dilation secondary to UVJ obstruction and that occurring secondary to high-grade vesicoureteral reflux (VUR). A clue to the diagnosis is variability in the diameter of the renal pelvis during one single examination, which favors a diagnosis of VUR. It is also important to distinguish the usually tortuous dilated ureter from loops of bowel. Another common cause of a dilated ureter is a ureterocele (discussed later).
The prognosis for UVJ obstruction is generally good, and the degree of ureteric dilation may offer some clue to the long-term prognosis and the need for postnatal surgery.
Duplex Collecting System/Ureterocele
Renal pelvic dilation secondary to a ureterocele or ectopic implantation of the ureter causes 5% to 7% of antenatal urinary tract dilation ( Fig. 15-14 ). Renal duplication can occur without dilatation, but is more readily demonstrated once dilation has developed ( Fig. 15-15 ). Most commonly the ureter draining the upper pole moiety has an ectopic insertion located inferior and medial to the normal bladder insertion site. Often an ectopic ureterocele is also seen within the bladder ( Fig. 15-14C ). Dilatation of the lower pole moiety most commonly occurs because of reflux. Duplex kidneys are thought to arise from an additional ureteric bud from the mesonephric duct.


A ureterocele is visualized as a septum or cyst within the bladder, although the ectopic extravesical insertion may be difficult to diagnose in utero ( Fig. 15-14C ). In some cases, the renal parenchyma of the obstructed side may be thinned and dysplastic. Although most cases of ureterocele are visible on prenatal ultrasound examinations, sonologists should be aware that a full bladder may sometimes compress the cyst and prevent visualization. A ureterocele may also prolapse into the urethra and result in acute LUTO.
The differential diagnosis of ureterocele includes UVJ obstruction and VUR. It should be remembered that 50% of duplex collecting systems have coexisting VUR in the lower pole moiety. The prognosis is generally good but will depend on the degree of associated renal dysplasia affecting each pole. In the prenatal period, follow-up ultrasound examinations to document worsening urinary tract dilation are indicated. Postnatal evaluation with further testing for residual renal function and reflux is recommended.
Vesicoureteral Reflux
VUR constitutes 10% to 20% of all diagnosed cases of antenatal urinary tract dilation. This diagnosis can be challenging to make prenatally and should be suspected in the presence of varying degrees of renal pelvis dilation with an associated either unilateral or bilateral dilated ureter ( Fig. 15-16A and B ). Proposed causes for VUR include transient bladder outlet obstruction that resolves prior to delivery, high voiding pressure resulting in distortion of the vesicoureteric junction, and delayed maturation of the vesicoureteric junction. There also appears to be a strong familial predisposition, raising the possibility of a genetic cause in some cases.

The diagnosis of VUR is important to confirm, as up to 40% of affected infants will develop renal scarring and long-term damage if undetected. The finding of a normal postnatal renal ultrasound examination does not exclude the diagnosis of VUR, and a micturating cystourethrogram may be indicated to completely rule out the diagnosis.
Lower Urinary Tract Obstruction
The most common cause of severe bladder outlet obstruction is a posterior urethral valve (PUV) ( Fig. 15-17 ). PUVs are membranes within the posterior aspect of the urethra. This condition affects male fetuses and typically presents with an enlarged bladder and dilated urethra, producing the classic keyhole sign seen on ultrasound imaging ( Figs. 15-17A and 15-18 ). The ureters and renal pelves are usually also dilated. With worsening obstruction, the bladder wall becomes hypertrophied with development of trabeculations. Renal parenchymal destruction and dysplasia may result from back-pressure, resulting in atrophic changes. Other findings in severe cases include severe oligohydramnios, perinephric urinomas, and urinary ascites ( Fig. 15-19 ).



The oligohydramnios seen in LUTO can result in phenotypic changes in the newborn, including Potter facies and contractures of the extremities. The most important consequence of oligohydramnios is the development of pulmonary hypoplasia, a significant contributor to infant mortality and morbidity risks.
Prenatal management involves differentiating bladder enlargement secondary to PUV from other causes of LUTO and from causes of nonobstructive bladder enlargement ( Table 15-4 ). The finding of massive bladder distention associated with oligohydramnios in a male fetus strongly suggests PUV. Careful assessment can identify fetuses with potential to benefit from prenatal intervention.
| Posterior urethral valves |
| Anterior urethral valves |
| Urethral atresia |
| Congenital megalourethra |
| Prolapsed ureterocele |
| Megacystis megaureter (vesicoureteral reflux) |
| Megacystis-microcolon–intestinal hypoperistalsis syndrome |
| Cloacal malformation |
Features of poor prognosis in cases of LUTO include early diagnosis, bilateral marked dilatation, persistently obstructed bladder, oligohydramnios, and secondary lung hypoplasia. Bilateral renal dilatation and LUTO have an increased risk of associated chromosomal anomalies, and, therefore, evaluation of fetal chromosomes may be warranted. The finding of associated hyperechogenic and cystic renal parenchyma is frequently associated with poor renal outcomes, with a positive predictive value of 59%, and negative predictive value of 56% regarding postnatal renal function. Conversely, although the presence of renal cortical cysts suggests a poor prognosis, normal renal cortical echogenicity does not exclude dysplasia. A systematic review including 13 articles and 215 women showed amniotic fluid volume and renal cortical appearance on ultrasound imaging to have only modest screening accuracy for predicting postnatal outcome ( Table 15-5 ).
| Criteria | Sensitivity | Specificity | AUC |
|---|---|---|---|
| Oligohydramnios | 0.63 (0.51-0.74) | 0.76 (0.65-0.85) | 0.74 |
| Renal cortex appearance | 0.57 (0.37-0.76) | 0.84 (0.71-0.94) | 0.78 |
| Gestational age at diagnosis <24 weeks | 0.48 (0.26-0.70) | 0.82 (0.66-0.92) | 0.68 ( P = 0.14) |
The role of measuring urinary electrolytes in the fetal urine obtained through transabdominal vesicocentesis to predict renal function is controversial. The process involves serial drainage of the fetal bladder with urine sent for biochemical analyses ( Table 15-6 ). The initial urine sample obtained may not be reliable for prognosticating renal function as it may have been stagnant over a long time. Furthermore, there are discrepancies in the predictive value of urine biochemistry in published studies on LUTO owing to small sample size, variations in cutoff values used, gestational age at interrogation, and sampling frequency in published studies. Fetuses with renal damage show decreased urinary concentrating ability, especially of sodium and calcium. Measurements of β 2 -microglobulin in the fetal urine has been suggested as demonstrating a better predictive accuracy, but a systematic review including 23 articles and 572 women found this protein to be less accurate than measurement of urinary sodium and calcium.
| Features | Levels/Findings |
|---|---|
| Sodium | <100 mg/dL |
| Chloride | <90 mg/dL |
| Osmolality | <200 mg/dL |
| Calcium | <8 mg/dL |
| β 2 -Microglobulin | <4 mg/dL |
| Total protein | <20 mg/dL |
| Amniotic fluid volume | Normal |
| Renal cortex appearance | Not cystic or echogenic |
| Gestational age at diagnosis | >24 weeks |
The goals of prenatal intervention are to avoid further renal damage and to prevent pulmonary hypoplasia. Vesicoamniotic shunting to divert urine from the obstructed fetal bladder into the amniotic cavity has been the primary prenatal intervention used for several years. However, the outcomes following vesicoamniotic shunting and the long-term results have not been convincing. The only randomized controlled trial evaluating this technique (the PLUTO [Percutaneous shunting in Lower Urinary Tract Obstruction] trial) was stopped early because of poor recruitment. Although the trial suggested a trend toward improved survival in the group that received shunting, the relative risk (RR) was not statistically significant (RR 1.88; confidence interval [CI] 0.71-4.96). The PLUTO trial was limited by a significant number of crossovers in the treatment arms; therefore, the authors performed an “as treated” analysis, which showed significant survival benefit from vesicoamniotic shunting. The long-term outcomes from the study are still pending.
Recent case reports have suggested the feasibility of fetoscopic placement of transurethral stents in fetuses with bladder outlet obstruction, although at this time the series are too small for reliable information on outcomes.
Urethral Atresia
Urethral atresia has a sonographic appearance similar to that of PUV and should be considered in female fetuses with bladder outlet obstruction. Unlike PUV, the obstruction is usually complete and may result in megacystis detected during the first trimester ( Fig. 15-20 ). The condition often has a poor prognosis.

Congenital Megalourethra
Congenital megalourethra is a rare condition affecting male infants, and it is usually associated with a dilated and elongated penile urethra that may appear sausage-shaped in continuity with a distended bladder ( Fig. 15-21A through C ). The proposed cause is delayed apoptosis at the penile meatus; this has also been referred to as “anterior urethral valves,” although the condition is not actually caused by valves. This process can lead to abnormal development or hypoplasia of the penile erectile tissue. Other sonographic features are similar to those seen in PUV. In some cases, the obstruction resolves spontaneously and the outcome can be favorable.

Cloacal Malformation
Cloacal malformation is a rare cause of bladder outlet obstruction affecting 1 in 50,000 pregnancies. It is caused by failure of development of the urorectal fold that usually separates the rectum from the genital tract. Consequently, a convergence of the gastrointestinal and genitourinary tracts occurs, with a common opening in the perineum. The condition is seen predominantly in females and can result in the collection of urine in the uterus and vagina (hydrometrocolpos) as well as bladder outlet obstruction and dilated ureters ( Fig. 15-22 ). Because of the complex embryologic anomalies involved, cloacal malformation can present with multiple phenotypes and associated anomalies including ureteral ectopia, bladder duplication, renal agenesis, sacral agenesis, open neural tube defects, and vertebral anomalies. The finding of ultrasound features suggestive of PUV in a female fetus should increase the suspicion for cloacal malformations. Transient ascites may be present from chemical irritation of the peritoneum by urine. It may be followed by intra-abdominal calcifications. Depending on the degree of bladder outlet obstruction, profound oligohydramnios may be present.

Prune-Belly Syndrome
Prune-belly syndrome (PBS) refers to an abnormally lax abdominal wall secondary to extensive stretching during early development. Historically, the condition was thought to result from primary partial or complete absence of the abdominal muscles, although it is currently thought that it is more likely a secondary result of overdistention due to bladder dilation that, in some cases, may have resolved spontaneously. The ultrasound features are very similar to those of PUV, but in PBS, the bladder distention and urethral dilation are not as marked. In addition, similar to PUV the condition is more common in male fetuses; therefore, diligent evaluation is required to distinguish these anomalies, as the therapeutic implications are significantly different prenatally. The prognosis is often poor, similar to that of PUV, secondary to severe pulmonary hypoplasia if oligohydramnios was present.
Megacystis-Microcolon–Intestinal Hypoperistalsis Syndrome
Megacystis-microcolon–intestinal hypoperistalsis syndrome (MMIHS) is an autosomal recessive condition occurring more commonly in females. It presents with evidence of a functional small bowel obstruction, a microcolon with intestinal malrotation, a markedly enlarged bladder, and bilateral hydronephrosis. The bladder finding is not secondary to a physical obstruction, as the condition is characterized by normal amniotic fluid volume and even polyhydramnios in late pregnancy. The underlying cause is poorly understood, but myogenic, neurogenic, and even hormonal mechanisms have been proposed. There may be associated anomalies of other organ systems, including omphalocele, cleft lip/palate, and cardiac defects, and the condition is associated with a poor prognosis that becomes apparent in the newborn period. The finding of an enlarged bladder with normal amniotic fluid volume suggests the differential diagnosis of MMIHS after excluding cloacal malformation.
Management of Fetal Renal Pelvic Dilation After Birth
Dilation of the renal pelvis is often the initial finding in abnormalities of the fetal urinary tract. Although there is generally a direct relationship between fetal renal pelvis dilation and obstructive processes of the urinary tract, there is considerable overlap with nonobstructive disorders. It should be appreciated that renal pelvis dilation detected prenatally often resolves spontaneously after birth with no correction required. In a series from our institution, 140 of 342 (40.9%) fetuses with renal pelvic dilation had nonobstructive hydronephrosis. In these cases, renal pelvis dilation detected prenatally was confirmed postnatally, but ultimately regressed without therapy.
Similarly, in a study of 350 infants with antenatally detected urinary tract abnormalities born in two Nottingham teaching hospitals in the United Kingdom, 170 of 350 (48.6%) were classified as “non-specific dilatation” (NSD). The majority of these (133/170) were categorized as NSD1. NSD1 referred to those fetuses with a renal pelvic diameter less than 10 mm in the AP diameter, no caliectasis or hydroureter, and normal renal cortex and size on renal ultrasound examination performed 4 to 6 weeks after birth. Infants in this group were exempt from further follow-up unless presenting with signs or symptoms of a urinary tract infection. Infants in the NSD2 group were those with a renal pelvis greater than 10 mm in the AP diameter. These infants subsequently underwent a voiding cystourethrogram (VCUG) or radionuclide renal scan to rule out reflux and renal drainage abnormalities, respectively. If normal, renal ultrasound examination was repeated within the first year of life. If renal pelvic dilatation was less than 10 mm, these infants were discharged from care with no further evaluation unless they developed symptoms of a urinary tract abnormality. The authors concluded that a large percentage of fetuses with renal pelvic dilatation can be served by a single postnatal renal ultrasound examination and that expectant management is appropriate and operative intervention is often unnecessary.
It is important that information relevant for the proper postnatal management of the newborn be transmitted to the pediatric team. In the majority of cases, no significant disease is observed postnatally, and the prenatal dilation represents transient or physiologic hydronephrosis. However, a variety of findings seen in the neonate after birth may suggest an abnormality of the urinary tract ( Fig. 15-23 ). After birth, some conditions require immediate confirmation and therapeutic maneuvers in order to protect renal function and prevent problems such as urinary tract infection, stone formation, and pain. These conditions include obstructive PUVs or the presence of an ectopic ureterocele that has prolapsed into the urethra, leading to oligouria or anuria and necessitating immediate treatment.

However, extensive evaluation is not always necessary and, in addition to time and expense, can result in discomfort and radiation exposure, as well as the consequences of false positive diagnoses and stress for the family. There is therefore some controversy as to which infants require radiologic evaluation and prophylactic antibiotics. In some cases, a workup should be planned but can be done on a less urgent basis. Ultrasound imaging is the least invasive and most common method of postnatal imaging. However, ultrasound examinations can only provide anatomic, not functional, evaluation, and findings are altered by bladder filling and hydration status. Because infants are relatively dehydrated at birth, later ultrasound evaluation (on the second day of life or later) is more accurate in assessing renal tract dilation. A normal ultrasound examination in the week after birth is not sufficient to verify the absence of disease and therefore later follow-up is often recommended ( Fig. 15-24 ).

Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


