Introduction
The old adage that “children are not young adults” is certainly true when evaluating pediatric head and neck lesions. The differential diagnosis of neck masses differs compared to masses that arise in adults. The most common pediatric head and neck lesions are congenital or developmental in origin, followed by inflammatory processes and tumors. This chapter will focus on the congenital/developmental and neoplastic processes; inflammatory lesions are covered in other chapters.
Benign Congenital Masses of the Neck
Thyroglossal duct cysts (TDCs) and anomalies of the branchial apparatus are the most common benign masses of the neck in children. Less common congenital benign masses of the head and neck in the pediatric age group include dermoid/epidermoid cysts and teratomas.
Thyroglossal Duct Cyst
TDC is the most common congenital neck mass and accounts for nearly 90% of nonodontogenic congenital cysts. TDC is the second most common benign cervical mass in children after reactive adenopathy and almost three times more common than branchial cleft cysts.
Embryogenesis of Thyroglossal Duct.
At about 3 weeks’ gestation, the thyroid gland begins to develop as a midline endodermal diverticulum from the floor of the pharynx, and over the following 4 weeks of development, the primitive thyroid gland enlarges to form a bilobed diverticulum. For a short period of time, the developing thyroid gland is connected to the tongue by a narrow tube, the thyroglossal duct. This duct courses from the region of the junction of the anterior two thirds and posterior two thirds of the tongue (foramen cecum) to the hyoid bone and farther caudally to the region of the future location of the pyramidal lobe of the thyroid gland in the inferior neck. A TDC or fistula forms when a portion of this duct fails to involute and leaves behind rests of secretory epithelial cells that when stimulated by an inflammatory process cause a cyst to develop.
Presentation.
The TDC is usually a midline or near-midline lesion and can occur anywhere along the path of the duct. About 20% to 25% occur in the suprahyoid neck, 15% to 50% at the level of the hyoid bone, and 25% to 65% in the infrahyoid neck. They commonly occur near the hyoid bone and can be superior, anterior, inferior, or posterior to this bone.
TDCs usually present in the first 5 years of life, with about 66% noted before age 7 and 90% before age 10. A small percentage are diagnosed in patients older than 50 years. Patients typically present with a history of a gradually enlarging mass in the midline or paramedian neck and demonstrate a nontender, mobile, 2- to 4-cm subcutaneous midline or paramedian neck mass of variable firmness. Other presentations include complications of cyst infection, fistula formation, or rupture.
The lining of the cyst may be stratified squamous, pseudostratified ciliated, simple cuboidal, or columnar epithelium, and there may be residual thyroid tissue in the enlarging mass in the midline of the neck. Coexisting carcinoma in the wall of the cyst is reported in less than 1% of patients with TDC and typically occurs in adults, with papillary carcinoma being most common followed by papillary-follicular carcinoma.
Imaging Findings.
Suprahyoid neck cysts are almost always in the midline. When the cysts occur just caudal to the hyoid bone at the level of the thyrohyoid membrane of the larynx, they can stretch this membrane and bow it posteriorly, appearing to lie in the preepiglottic space of the larynx. In the infrahyoid neck, the cyst lies just off the midline, adjacent to the outer surface of the thyroid cartilage and deep to the infrahyoid strap muscles.
In the pediatric population, initial imaging evaluation is frequently performed with ultrasound, which demonstrates a well-defined, thin-walled, midline or paramedian mass with increased through transmission and variable internal echogenicity and no internal blood flow. Increased internal echogenicity can be due to multiple factors including inflammatory debris, hemorrhage, or proteinaceous content.
On computed tomography (CT), the TDC appears as a mass of relatively decreased attenuation to muscle. When the cyst is not infected, it will also demonstrate a thin, smooth rim, but when infected the cyst wall thickens and enhances ( Fig. 26-1 ). The attenuation of the cyst will vary based on its contents; usually they will have mucoid attenuation (10-25 Hounsfield units [HU]), but if there has been previous infection or hemorrhage, attenuation of the cyst contents can approach that of muscle. On magnetic resonance imaging (MRI) the T1 signal intensity can vary from low to high, while T2 signal intensity remains high. The variations in signal intensity are based on the variable protein content of the cyst. With infection or hemorrhage, septations can also arise in the cyst.

A TDC with an internal eccentric solid mass containing calcifications or infiltrative soft tissue characteristics should raise concern for an associated carcinoma.
The primary differential considerations of TDC by imaging include dermoid/epidermoid cyst, branchial cleft cyst (if paramedian in location), or rarely a sebaceous cyst.
Treatment.
Treatment is surgical removal of all tissue along the course of the duct as far as the foramen cecum (Sistrunk procedure). The hyoid bone rotates during maturation before assuming its final adult position. During this rotation, the thyroglossal duct is adherent to the hyoid along its anterior inferior edge and may be drawn posteriorly and cranially to lie behind the body of the hyoid bone; rarely the duct can be incorporated into the hyoid bone, trapped between the second and third arch components of the hyoid’s body. Owing to this close relationship between the body and the TDC, Sistrunk proposed that the body of the hyoid bone should be removed during surgical resection of a thyroglossal duct, leading to a significant decrease in recurrences, reduced from nearly 50% to less than 4%.
Anomalies of the Branchial Apparatus
Embryogenesis of the Branchial Apparatus.
The anatomic structures of the face and neck predominantly derive from the branchial apparatus, which is a complex structure derived from neural crest cells that develops between the second and seventh weeks of gestation. The branchial apparatus consists of six paired arches separated on their outer surface by five paired ectodermal clefts and on their inner surface by five paired endodermally derived pharyngeal pouches. By the end of the fourth week of life, four well-defined pairs of arches are visible and the fifth and sixth arches are rudimentary. Each arch is composed of a central core of mesoderm and is lined externally by ectoderm and internally by endoderm.
The cervical sinus of His is formed after the branchial arches appear by accelerated mesodermal growth cranially of the first arch and a portion of the second arch, and caudally by growth of the epipericardial ridge, which develops from the mesoderm lateral to the fifth-sixth arch. The branchial apparatus typically disappears between the fourth and sixth weeks of life.
Etiology of Branchial Anomalies.
The etiology of branchial anomalies is controversial, and the most accepted theories to explain the development of branchial abnormalities propose that they are either vestigial remnants from incomplete obliteration of the branchial apparatus or the result of buried epithelial cell rests.
Classification.
Anomalies of the branchial apparatus are best understood by being classified into a spectrum of developmental anomalies that includes fistulas, sinuses, and cysts.
First Branchial Anomalies.
First branchial cleft anomalies account for up to 5% to 8% of all branchial anomalies: 68% are cysts, 16% are sinuses, and 16% are fistulas. Although Arnot and Work tried to subclassify first branchial anomalies into two distinct subtypes in 1971 and 1972, Olsen—after reviewing multiple cases—concluded that it is difficult to subclassify all these anomalies into one of the subtypes and proposed a simplified classification (cysts, sinuses, and fistulas).
Embryology.
The first branchial apparatus gives rise to portions of the middle ear, external auditory canal, eustachian tube, mandible, and maxilla, and the formation of these structures is completed by the sixth and seventh weeks of gestation. The parotid gland and facial nerve form and migrate between the sixth and eighth weeks of gestation. Because the facial nerve and parotid gland have a somewhat later embryologic development, vestigial branchial anomaly can be located in variable relationship to the parotid gland and the facial nerve. First branchial apparatus anomalies arise from incomplete closure of the ectodermal portion of the first branchial cleft. They can start at the junction of the bony and cartilaginous external auditory canal or in the cartilaginous portion of the external canal, the plane between the mandibular and hyoid arches, and end in the submandibular area. As a result, first branchial apparatus anomalies can arise in the middle ear cavity, external auditory canal, superficial or deep lobes of the parotid gland, superficial to the parotid gland, at the angle of the mandible, anterior or posterior to the pinna, or along the nasopharynx.
Presentation.
Although the majority of first branchial apparatus anomalies occur in the first decade of life, they can also be encountered in middle-aged patients, especially women. Patients typically present with recurrent abscesses or inflammatory processes with tenderness and swelling at the angle of the mandible or in the ear. If the process is related to the parotid gland, patients tend to present with recurrent parotid abscesses not responding to therapy. If there is drainage of the cyst into the external auditory canal, the initial presentation is typically otorrhea. Simple cysts can also present as a painless soft tissue mass. Sinus tracts are rare with first branchial anomalies. When present, they are usually found in the first decade of life.
Imaging findings.
First branchial cysts are usually related to the parotid gland, external canal of the ear, and/or the lower margin of the pinna or at the angle of the mandible. If a tract can be identified, directed toward the external auditory canal, then the diagnosis can be made with certainty. If a tract is not seen and the cyst lies within the parotid gland, possibilities would include a first branchial cyst, parotid suppurative lymph node and abscess, inflamed ectatic salivary gland, a lymphoepithelial cyst (human immunodeficiency virus [HIV]), Sjögren’s syndrome, or a rare localized obstructive mucocele.
Uncomplicated cysts demonstrate mild rim enhancement or no enhancement. Infected cysts have variable wall thickness and enhancement characteristics ( Fig. 26-2 ). If the anomaly is located within the parotid gland, the imaging findings will be nonspecific. For example, the cyst contents of a first branchial cyst, a lymphoepithelial cyst, or an obstructive mucocele or sialocele are usually of mucoid attenuation on CT, and on MRI usually have low to intermediate T1-weighted and high T2-weighted signal intensity. T2-weighted or short tau inversion recovery (STIR) MRI sequences may be helpful in identification of small fluid-filled tracts leading to the external auditory canal. If the cyst is infected and has a tract, coronal T2-weighted and postcontrast T1-weighted fat-suppressed MRIs would best demonstrate the tract.

Treatment.
Treatment is complete surgical resection and carries an excellent prognosis. The major complication of surgery is related to facial nerve injury. Residual wall remnants can lead to recurrence.
Second Branchial Anomalies.
About 95% of all branchial anomalies are related to the second branchial apparatus, and these anomalies can occur anywhere from the tonsillar fossa to the supraclavicular region of the neck. The majority of these anomalies, about three fourths, are cysts. Cysts are more common between 10 and 40 years of age, and fistulas or sinuses usually present before age 10.
Classification.
Second branchial cysts are classified into four subtypes based on location per Bailey’s classification. Type 1 cyst lies deep to the platysma muscle and anterior to the sternocleidomastoid (SCM) muscle ( Fig. 26-3 ). This type of cyst is felt to represent a remnant of the tract between the sinus of His and the skin. Type II cyst is the most common type and is thought to be the result of the persistence of the sinus of His. This cyst is located posterior and lateral to the submandibular gland, anterior and medial to the SCM muscle, and anterior and lateral to the carotid space ( Fig. 26-4 ). Type III cyst/fistula is thought to arise from a dilated pharyngeal pouch and courses medially between the internal and external carotid arteries; it can extend up to the lateral wall of the pharynx or skull base ( Fig. 26-5 ). Type IV cyst lies in the mucosal space of the pharynx adjacent to the pharyngeal wall and is thought to arise from a remnant of the pharyngeal pouch ( Fig. 26-6 ).




Presentation.
The most common presentation of a second branchial cleft cyst is as a fluctuant nontender mass at the lateral aspect of the mandibular angle. When infected the patient will present with a history of a slowly enlarging painful mass. Because the cysts contain lymphoid tissue, stimulus such as an upper respiratory tract infection can lead to an increase in size of the cyst. With a fistula, an ostium is often noted at birth at the anterior border of the junction of the middle and inferior thirds of the SCM muscle. The tract then courses deep to the platysma muscle, ascends laterally along the carotid sheath, lateral to the hypoglossal and glossopharyngeal nerves, and then passes between the internal and external carotid arteries before it terminates in the region of the palatine tonsillar fossa.
Imaging findings.
Ultrasound is usually the initial imaging study because the cysts often present as a neck mass in a young child and can range in size from 1 to 10 cm. If the anomaly is uncomplicated it will demonstrate sonographic characteristics of a simple cyst (thin wall, increased through transmission, anechoic, and compressible). On CT the anomaly will demonstrate a thin wall with decreased attenuation. On MRI it will appear as hypointense to slightly hyperintense to muscle on T1-weighted images and hyperintense to muscle on T2-weighted images. When it is infected it can demonstrate a thicker wall with increased internal echogenicity on ultrasound, increased attenuation on CT, and hyperintense T1-weighted signal on MRI, with variable enhancement. If the cysts are chronically complicated, they can demonstrate septations.
Sometimes there may be a “beak sign” present, which is pathognomonic of a Bailey type III second branchial cyst, where the medial aspect of the cyst is compressed and forms a beak as it extends between the internal and external carotid arteries on axial CT or MRI.
In children, differential considerations include suppurative adenopathy/deep neck infection, lymphatic malformation, dermoid/epidermoid cyst, and thymic cyst. Other rare considerations in this age group include cystic schwannoma of the vagal nerve, minor salivary gland tumor, and necrotic malignant adenopathy. Because the jugulodigastric node is seen in the same location as the most common form of second branchial cleft cyst, clinicians may mistake it for an enlarged, suppurative, reactive, or tumor-infiltrated jugulodigastric node.
Treatment.
Treatment is complete surgical resection, which carries an excellent prognosis.
Third and Fourth Branchial Anomalies.
These are rare anomalies, and third branchial anomalies account for 3% and fourth branchial anomalies account for about 1% to 2%. Third branchial anomalies are centered in the posterior cervical space, and despite their overall rarity they are the second most common congenital lesions of the posterior cervical space after lymphatic malformation. These must sometimes be distinguished from the large second branchial cleft cyst, which can protrude posteriorly in the posterior cervical space.
Embryology.
Third branchial anomalies most likely arise secondary to failure of involution of the third branchial apparatus, and fourth branchial anomalies are likely due to failure of regression of the fourth branchial pouch or distal sinus of His.
Third branchial apparatus cysts are located posterior to the common carotid or internal carotid artery, between the hypoglossal nerve (below) and glossopharyngeal nerve (above). If it is in the form of a fistula, there will be a cutaneous opening similar to a second branchial fistula that is anterior to the lower SCM muscle. It then courses posterior to the common or internal carotid artery, anterior to the vagus nerve and between the hypoglossal and glossopharyngeal nerves, and then pierces the thyrohyoid membrane and enters the pyriform sinus anterior to the superior laryngeal nerve.
Presentation and imaging findings.
Third branchial cleft cysts usually present as painless, fluctuant, 2- to 5-cm masses in the posterior triangle of the neck, often after a viral upper respiratory infection ( Fig. 26-7 ). The presentation is at an earlier age with a sinus or a fistula. On CT and MRI, uncomplicated cysts usually present as a unilocular cystic mass on imaging, and complicated cysts demonstrate increased wall thickness with variable enhancement characteristics and internal proteinaceous fluid.

Anomalies related to the fourth branchial pouch are usually in the form of a sinus tract that arises from the pyriform sinus, pierces the thyrohyoid membrane, descends along the tracheoesophageal groove, and continues into the mediastinum. These anomalies have variant presentations such as a nontender fluctuant mass anterior to the inferior SCM muscle for uncomplicated cases, and suppurative thyroiditis or recurrent neck abscesses for complicated cases ( Fig. 26-8 ). For diagnosing a fistulous tract by imaging, a contrast-enhanced CT following ingestion of barium or water-soluble contrast or direct injection of a fistulous tract is the imaging study of choice.

Although it is difficult to differentiate third from fourth branchial anomalies by imaging alone, the key anatomic markers for correctly diagnosing them include the carotid artery and the superior laryngeal nerve. Third branchial anomalies lie posterior to the internal carotid artery and above the superior laryngeal nerve, whereas fourth branchial anomalies are anterior to the internal carotid artery and lie below the superior laryngeal nerve. Third and fourth branchial anomalies can be related to the pyriform sinus and can appear similar to external laryngoceles on imaging.
Other differential considerations for third and fourth branchial anomalies include other branchial anomalies, TDC, lymphatic malformation, thyroid abscess, thyroid, parathyroid, or thymic cyst, and suppurative adenopathy.
Treatment.
For both third and fourth branchial anomalies, complete surgical resection is the treatment of choice. Persistence of the tract to the pyriform sinus can result in recurrent episodes of thyroiditis and can necessitate a partial thyroidectomy. If fourth branchial anomalies are complicated by infection and secondary thyroiditis, medical treatment should precede surgical treatment.
Cervical Thymic Remnant
Embryology and etiology.
Cervical thymic remnants are very rare lesions, and two main theories, congenital theory and acquired theory, have been reported to explain their etiology. The congenital theory states that the cysts are due to persistence of thymopharyngeal duct remnants and can be located anywhere from the level of the pyriform sinus to the superior mediastinum and located either beneath or medial to the SCM muscle. The acquired theory for thymic cysts proposes progressive cystic degeneration of thymic (Hassall’s) corpuscles, primitive endodermal cells, lymphocytes, and epithelial reticulum of the thymus as sources for thymic remnants.
Presentation.
The thymus reaches its greater relative size at age 2 to 4 years and its greatest absolute size at puberty. Cervical thymic remnants are very rare lesions and can be found anywhere along the path of the thymopharyngeal duct.
Most are detected in childhood, and two thirds are found in the first decade of life, often between 3 and 8 years of age, and the remainder in the second and third decades of life.
Most patients are asymptomatic, and about 80% to 90% present with a painless slowly enlarging neck mass near the thoracic inlet, anterior or deep to the SCM muscle. In 50% of cases, a connection between the cervical thymic anomaly and the mediastinal thymic gland occurs in the form of direct extension or a remnant of the thymopharyngeal duct. Mediastinal thymic cysts can occur without a cervical component. There is a frequent association between cervical thymic cysts and thyroid and parathyroid inclusion cysts, and a fibrous strand is sometimes present between the cervical thymic cyst and the thyroid gland.
Imaging findings.
Because most ectopically located thymic masses are often cystic on ultrasound, CT and MRI will demonstrate imaging characteristics consistent with an uncomplicated cyst. A cystic lesion will course parallel to the SCM muscle and along the carotid sheath or lateral aspect of the visceral space on CT ( Fig. 26-9 ). Imaging characteristics will vary if the cysts are complicated by hemorrhage, cholesterol, proteinaceous fluid, or infection. Aberrant thymic tissue, parathyroid tissue, or lymphoid tissue can be present as soft tissue components related to the wall of the cyst.

Differential diagnosis.
A second branchial apparatus fistula is the main differential consideration of thymic remnants. A second branchial fistula will pass between the internal and external carotid arteries and end in the superior tonsillar pillar, whereas a thymic cyst will pass posterior to the carotid bifurcation and terminate in the pyriform sinus. Additionally, about 50% of cervical thymic cysts extend into the superior mediastinum, and some can be seen in the inferior pole of the thyroid gland. Other differential considerations for a thymic cyst include a fourth branchial anomaly, TDC, thyroid/parathyroid cyst, lymphatic malformation, cyst neuroblastoma, lymphadenopathy, external laryngocele, and vallecular cyst.
Treatment.
Treatment is surgical, and the prognosis is excellent if the resection is complete. There is a high recurrence rate with incomplete resection.
Dermoid/Epidermoid Cyst
Because of overlapping features, the terminology and classification of dermoid, epidermoid, and teratoid cysts in the head and neck can lead to confusion.
Embryology.
These masses are thought to arise from trapped epithelial cell rests at the site of midline closure. Dermoids and epidermoids are ectodermal-lined inclusion cysts, and teratoids can contain tissues of all three germ-cell layers. Dermoids contain epithelial and dermal elements, whereas epidermoids contain only epithelial elements.
Presentation.
There is no gender predilection with dermoid and epidermoid cysts. Dermoid cysts of the neck commonly occur near the midline, with the most common location being the floor of the mouth, (11.5% of all dermoids), followed by other locations including the anterior neck, tongue, palate, and orbit. Epidermoid cysts are rarely found in the head and neck, and when they occur they usually present in infancy.
Dermoid/epidermoid cysts become clinically apparent in the second or third decades of life, when they present as a painless, soft, slow-growing mass in the suprahyoid midline neck. The cyst can be variable in size, and rapid increase in size can be seen secondary to association with a sinus tract, pregnancy, or increasing desquamation of skin appendage products. Masses lying in the sublingual space are often clinically inapparent and demonstrate minimal mass effect, but masses in the submandibular space present with a more obvious swelling or mass effect. Additionally, a distinguishing factor between TDCs and dermoid/epidermoid cysts are that whereas TDCs move with protrusion of the tongue, owing to their association with the hyoid bone, dermoid/epidermoid cysts are nonmobile with this maneuver.
Dermoid cysts are well-defined encapsulated masses lined with squamous epithelium and may contain skin appendages in the form of sweat or sebaceous glands or hair follicles, which can lead to the formation of keratin and sebaceous material, including occasional hair. Epidermoid cysts lack skin appendages but have a histologic appearance similar to dermoids. The incidence of malignant degeneration into squamous cell carcinoma is about 5% for dermoids.
Imaging Findings.
On CT a dermoid cyst can appear as a thin-walled unilocular mass with decreased attenuation and at times with CT attenuation values approximating that of fat ( Fig. 26-10 ). Sometimes a virtually pathognomonic “sack-of-marbles” appearance, representing multiple small fatty nodules within the fluid, is seen within the cyst. These cysts can also present with a more heterogeneous appearance owing to differences in composition of the various skin appendage components, including calcification. Thin peripheral enhancement can be seen following contrast administration.

On MRI, signal changes vary depending on the composition of the various cyst components. T1 signal can be hyperintense due to lipid components or isointense to muscle, and T2 signal is usually hyperintense to muscle. If calcification is present, this will cause low T2 signal. If a dermoid contains minimal complex elements, it can appear similar to an epidermoid. Postcontrast images may or may not demonstrate rim enhancement. T1 postcontrast fat-saturation images can be helpful to diagnose dermoids by assessing for signal dropout.
Epidermoid cysts usually follow water signal, demonstrating homogenous low attenuation on CT and hypointense T1-weighted and hyperintense T2-weighted signal to muscle on MRI ( Fig. 26-11 ). The signal characteristics of this lesion can be confused with other cystic lesions such as an uncomplicated lymphatic malformation or ranula.

It is important to accurately localize the lesions when they are located in the floor of the mouth. If the lesion is located above the mylohyoid muscle, it lies in the sublingual space and is amenable to an intraoral surgical approach. If it lies in the submandibular space, a submandibular approach would be used. The optimal imaging plane for localizing these masses into the appropriate space would be coronal CT or MRI.
Teratoma
Approximately 9% of all pediatric head and neck neoplasms are teratomas, and less than 5% of all teratomas occur in the head and neck. In the pediatric population, most teratomas are extragonadal in location, with 82% in the sacrococcygeal area; locations in the head and neck include neck, nasal cavity, paranasal sinus, oral cavity, oropharynx, nasopharynx, temporal bone, ear, and orbit.
Teratomas are thought to arise from misplaced pluripotential primordial germ cells and are classified as true neoplasms because of their biological behavior with progressive and invasive growth patterns.
Cervical Teratomas.
Cervical teratomas are large and can present with extensive unilateral or more diffuse cervical swelling that can lead to obstetric complications such as difficult labor with malpresentation, premature labor, stillbirth, acute neonatal respiratory distress, and maternal polyhydramnios. The tumors that present later in life are smaller but have a higher incidence of malignancy.
Embryology.
Cervical teratomas can contain variable degrees of maturation, with mature elements derived from ectoderm, mesoderm, and endoderm and immature tissue mostly from neuroectoderm. Both mature and immature neuroectodermal elements tend to predominate in cervical teratomas.
Imaging findings.
On antenatal ultrasound, teratomas can appear well defined or infiltrative and demonstrate heterogeneous echogenicity with multiloculated areas of cystic and solid regions and acoustic shadowing from calcifications on sonography.
On CT, the imaging characteristics will reflect the pathologic makeup of the tumor. Most cervical teratomas are large, measuring up to 12 cm, and can appear primarily cystic, solid, or multicystic; about 50% contain calcification ( Fig. 26-12 ). On MRI, the mass shows heterogeneous signal on all sequences and contains areas of T1 hyperintense signal in tissues containing fatty components and hypointense or hyperintense signal in areas of calcification. Variable enhancement is seen after contrast administration. Because these tumors are large and can lead to airway compromise, intubation is often required.

Differential diagnosis.
The primary differential diagnosis is lymphatic malformation, which more closely follows water signal on all pulse sequences. Because cervical teratoma can be difficult to distinguish from a lymphatic malformation with prenatal ultrasound, fetal MRI has been found to be helpful in such cases. Other differential considerations include cervical neuroblastoma (which can contain calcification), complicated TDC or branchial cleft cyst, goiter, hemangioma, and external laryngocele.
Treatment.
For congenital cervical teratomas, surgery is mandatory and the mortality rate is 80% to 100% secondary to airway complication. Therefore differentiating this tumor from other congenital space-occupying masses of the neck is important. The incidence of true malignancy is less than 5%, and associated anomalies are uncommon. Prognosis with surgery is excellent.
Congenital Masses of the Nose
Nasal Dermal Sinus Cyst
Nasal dermal sinuses are thin epithelium-lined tubes that arise at external ostia situated along the midline of the nose and extend deeply for a variable distance, extending sometimes to the intracranial compartment. Nasal dermoid and epidermoid cysts are midline epithelium-lined cysts and may coexist with dermal sinuses or present as isolated masses.
Nasal dermal sinus cyst (NDSC) is the most common midline congenital lesion and accounts for 1% to 3% of all dermoids and 3.7% to 12.6% of all dermoids of the head and neck. Although most are sporadic, several familial cases have been reported. They can present at any age but are most commonly found in younger children, with a mean age of presentation of 3 years. There is a male predilection, and these lesions are found anywhere between the glabella and the base of the columella. About 56% present as midline cysts and 44% as midline sinus ostia.
Embryology.
Early in the embryo, the developing frontal bones are separated from the developing nasal bones by the fonticulus frontonasalis. The nasal bones are separated from the cartilaginous nasal capsule by the prenasal space, which extends from the base of the brain to the nasal tip. The most accepted theory for the development of NDSC is the failure of involution of the anterior neuropore, with lack of regression of the diverticulum of dura that extends through the foramen cecum. The midline diverticula of dura normally project anteriorly into the fonticulus frontonasalis and anteroinferiorly into the prenasal space and touch the ectoderm. These diverticula usually regress before closure of the bone plates of the anterior skull base, but if the embryonic diverticula of dura become adherent to the superficial ectoderm, they may not regress normally and instead may pull ectoderm with them as they retreat, creating a dermal/ectodermal tract that extends from the glabella through a canal at the frontonasal suture to the crista galli or beyond the crista to the interdural space between the two leaves of the falx. Depending on the contents and patency of the diverticulum, the resultant lesion can be a dermal sinus, dermoid cyst, nasal glioma, or encephalocele.
Presentation.
NDSC commonly presents as a painless cystic enlargement of any part of the nose, associated with one or more dimples. Externally, midline pits, fenestra, or discrete masses can be present, and they may be associated with hair protruding from the defect. With intermittent episodes of inflammation or discharge of sebaceous or inflammatory material there can be a variation in the size of the mass. Although there is no consistent associated pattern of malformation of the face and brain, an increased incidence of intracranial extension of the NDSC has been found in patients with multiple malformations.
The extension of nasal dermoid cysts and dermal sinuses is variable, which may end blindly in the superficial tissues or extend both intracranially and extracranially. The reported intracranial extension varies between 0% and 57%. Intracranial extension of the dermoids is typically extraaxial, with extension usually occurring through the foramen cecum into the epidural space in the region of the crista galli ( Fig. 26-13 ).

When intracranial extension is present, signs and symptoms of meningitis or behavioral changes related to life-threatening frontal lobe abscess can be seen. Other complications include nasal abscess, periorbital-nasal cellulitis, osteomyelitis, cavernous sinus thrombosis, and seizures.
Imaging Findings.
Both CT and MRI are useful in evaluating the course of the sinus tract, cysts, and sequelae of infection. On CT, the course of a dermal sinus is seen as a well-defined gap between the nasal bones in the midline or in the frontal bone. An enlarged foramen cecum and bifid-appearing or distorted crista galli are suggestive of but not pathognomonic for intracranial extension. These findings can present with or without intracranial extension or with intracranial extension of a fibrous cord without dermoid elements. The intracranial ends of dermoid cysts usually lie in a hollowed-out space along the anterior surface of a thickened enlarged crista galli. The ostium and tract usually appear as isodense fibrous channels or as lucent dermoid channels that can extend inward for variable distances. The bony canals indicate the course of the tract through the nasal bones, ossified nasal septum, and skull base.
On CT, noninfected dermoid cysts appear as nonenhancing masses of fat attenuation with an isodense rim to soft tissue. Stranding of the surrounding soft tissues and change in attenuation value of the cyst suggest secondary infection. An infected dermoid cyst has an appearance similar to an abscess, with a peripheral rim of enhancement. A foramen cecum can be incompletely ossified, a normal finding in children younger than 1 year of age, appearing as a bony defect. In such cases, contrast administration can aid in the differentiation of enhancing cartilage in a young child from a nonenhancing true skull base defect, similar to differentiation of enhancing nasal mucosa from nonenhancing dermoids.
MRI is extremely valuable in assessing the intracranial and nasal components of these lesions. Thin-slice sagittal, coronal, and axial T1-weighted, T2-weighted with fat saturation, and fast-spin echo inversion recovery sequences should be obtained. Sagittal T1-weighted and inversion recovery sequences are best for demonstrating enlargement of the foramen cecum with intracranial extension, and postcontrast imaging with fat saturation has similar appearances as CT. Dermoid cysts commonly have close to fat signal as compared to brain on all sequences, with associated chemical shift artifact and suppression with fat saturation. Fibrous tracts demonstrate an isointense signal to brain on T1-weighted sequences.
Treatment.
The nasal dermal sinuses are resected for three major reasons: for cosmetic purposes, to avoid/treat complications of local infection, and to avoid/treat secondary complications such as secondary meningitis and cerebral abscess. To avoid unnecessary craniotomies, surgical studies suggest that the best approach is to dissect the extracranial portion of the tract along its entire length from the superficial ostium to the extracranial surface of the enlarged foramen cecum and sever the tract at the extracranial end of the foramen cecum. Then send the severed end for pathologic examination, and if the specimen shows (epi)dermal elements at the foramen cecum, the dissection is extended intracranially. If no (epi)dermal elements are found at the foramen cecum, the procedure is concluded without intracranial exploration.
Nasal Gliomas (Heterotopias)
Nasal gliomas are congenital masses of glial tissue that may occur intranasally and/or extranasally at or near the root of the nose and may or may not be connected to the brain by a pedicle of glial tissue. They do not contain cerebrospinal fluid (CSF)-filled space that is connected with either of the CSF-containing spaces in the brain (ventricles or subarachnoid space). They are uncommon and account for only 4.5% of congenital nasal masses. They occur sporadically with no familial tendency and usually affect both genders equally. About 15% of patients with nasal heterotopias also manifest multiple cerebral heterotopias.
Nasal gliomas are subclassified into three types: extranasal (60%), intranasal (14%-30%), and mixed (10%-14%) forms. Extranasal gliomas lie external to the nasal bones and nasal cavities and typically occur at the bridge of the nose, to the left or right of the midline. Intranasal gliomas lie within the nasal or nasopharyngeal cavities. Mixed nasal gliomas consist of extranasal and intranasal components and communicate via a defect in the nasal bones or around the lateral edges.
Presentation.
Extranasal gliomas present in early infancy or childhood as firm, slightly elastic, reddish to bluish, skin-covered masses, and capillary telangiectasias may cover the lesion. They do not pulsate, do not increase in size with the Valsalva maneuver, and do not pulsate or swell following compression of the ipsilateral jugular vein. Intranasal gliomas lie within the nasal or nasopharyngeal cavities and present as large, firm, polypoid, submucosal masses that may extend inferiorly toward the nostril. They may protrude through the nostril and usually attach to the turbinates and come to lie medial to the middle turbinate between the middle turbinate and nasal septum. These can lead to obstruction of the nasal passage or blockage of the nasolacrimal duct, causing epiphora on the affected side. CSF rhinorrhea, meningitis, and epistaxis may be additional complications and presenting complaints.
Intranasal gliomas are commonly confused with inflammatory polyps, which usually lie medial to the middle turbinate, whereas inflammatory polyps typically lie inferolateral to the middle turbinate. Nasal gliomas usually present in infancy, whereas ordinary nasal polyps are rare in children younger than 5 years.
Histologically, nasal gliomas resemble reactive gliosis rather than neoplasia and consist of large aggregates or small islands of glial tissue. Fibrous connective tissue enwraps the blood vessels and extends outward to form septa that divide the mass. Extranasal gliomas are surrounded by dermis, and intranasal gliomas are surrounded by minor salivary glands, fibrovascular tissue, and nasal mucosa.
Imaging Findings.
On imaging, nasal gliomas and encephaloceles appear as masses of soft tissue signal isointense to gray matter. The differentiation between a nasal glioma and an encephalocele is made based upon identification of intracranial communication. If an intracranial communication is identified, the mass is classified as an encephalocele. This is best evaluated with MRI in the sagittal plane.
Cephaloceles
Cephaloceles are defined as herniations of intracranial contents through a cranial defect. When the herniation contains only meninges, it is defined as a cranial meningocele, and if the herniation contains brain, then it is called a meningoencephalocele. They are classified by the site of the cranial defect through which the brain and meninges herniate. Cephaloceles are common lesions, occurring in 1 per 4000 live births. The types of cephaloceles vary based on different populations. Sincipital cephaloceles are found in 1 in 35,000 live births in North America and Europe but are more common in Southeast Asia, where they are found in 1 per 5000 to 6000 live births. In Caucasians, occipital cephaloceles are most common (67%-80%) whereas sincipital cephaloceles (2%-15%) and basal cephaloceles (10%) are uncommon. Sincipital encephaloceles commonly present in the neonatal or infantile period with a nasal mass or nasal stuffiness, and nasopharyngeal basal cephaloceles present toward the end of the first decade with mouth breathing or nasal stuffiness.
Anterior (Sincipital Cephaloceles).
Anterior cephaloceles, also known as sincipital cephaloceles, are situated in the anterior part of the skull and include both interfrontal and frontoethmoidal cephaloceles. They always present as external masses along the nose, orbital margin, or forehead. With the interfrontal cephalocele, the cranial defect lies between two frontal bones and the cephalocele presents anteriorly as a midline mass between the defect.
Frontoethmoid cephaloceles emerge outward from the skull defect at the junction of the frontal and ethmoid bones, immediately anterior to the crista galli. They are subclassified into nasofrontal, nasoethmoidal, and nasoorbital subtypes based on the point at which the skull defect and hernia emerge externally. Various complications can occur depending on the size of the mass. For example, large masses stretch and thin the skin, obscure vision, block the airway, and may also cause pressure deformities of adjacent soft tissue and bone at the forehead, nose, and orbits, causing telecanthus or true bony hypertelorism.
Basal Cephaloceles.
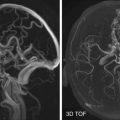
Basal cephaloceles are subdivided into transethmoid, transsphenoidal, sphenoethmoid, and frontospenoid. The most common type is the sphenoethmoid type, and it usually presents as a pharyngeal mass with signs and symptoms of airway obstruction or CSF leak ( Fig. 26-14 ). Other complications that can be seen with basal cephaloceles (transsphenoidal or sphenoethmoidal) include pituitary and hypothalamic dysfunction and decreased visual acuity secondary to herniation of the pituitary gland, hypothalamus, and optic apparatus into the sac.

Embryogenesis.
There is no universal agreement on the etiology of anterior cephalocele, but it is thought to be secondary to imperfect closure of the anterior neuropore of the neural tube at about the 25th day of gestation. The basal cephaloceles, those related to the sphenoid bone, are thought to be secondary to persistence of the craniopharyngeal canal or failure of normal union of basilar ossification centers.
Histology.
In a meningoencephalocele, the herniated neural tissue contains neurons with prominent astrogliosis, and the cyst wall is composed of glial and fibrous connective tissue without neurons.
Imaging.
CT is useful for imaging the bony margins of the skull base defect along with the soft tissue component of the lesion. MRI is the best imaging modality for evaluating the contents of the cephalocele and in defining the cephalocele in relation to the pituitary stalk and gland as well as the position and state of the optic nerves, chiasm, and optic tracts.
Nasopharyngeal Teratomas
Head and neck nasopharyngeal teratomas are presumed to arise from primitive germ cells that get trapped in the normal migratory channel during development. They contain all three germ layers and arise from embryonic tissue that breaks off or fails to migrate to its normal destination. More than 50% of nasopharyngeal teratomas are seen in the first year of life and often arise from the superior or lateral wall of the nasopharynx. There is a female predilection for teratomas in the nasal cavity, and the typical clinical presentation is a neonate with severe respiratory distress, stridor, and dysphagia. CT and MRI are the best imaging modalities for teratomas, which can be identified by the presence of fat and and calcification. Differential considerations for nasopharyngeal teratomas include dermoids, encephalocele, nasal glioma, rhabdomyosarcoma, hemangioma, lipoma, neurofibroma, and lymphoma.
Malignant Neoplasms
Neoplasms of the head and neck comprise approximately 5% of all malignant neoplasms in the pediatric age group. Although lymphoma and rhabdomyosarcoma are the most common tumors, other tumors in this category include primary neuroblastoma, nasopharyngeal carcinoma (NPC), thyroid carcinoma, synovial sarcoma, and esthesioneuroblastoma.
Lymphoma
Lymphoreticular malignancies account for about 40% of all malignant disorders in children, with lymphoma representing 11% of pediatric malignancies and 50% of pediatric head and neck malignancies. It is the second most common malignancy in the head and neck in this age group and is equally divided between non-Hodgkin’s and Hodgkin’s lymphoma.
Hodgkin’s Lymphoma.
Hodgkin’s lymphoma represents 14% of all cases of lymphoma and has a bimodal age distribution: the first peak in teenagers and young adults and the second peak in adults older than 40 years. The disease is uncommon in children younger than age 5, and there is a male predominance of 3 : 1 in the preteen years. Although the etiology remains unknown, there is a known association with Epstein-Barr virus in up to 50% of cases. There is also increased incidence of the disease in patients with HIV, which carries a poorer prognosis. The disease is less common among Asian and African American populations.
The majority of patients present with painless adenopathy, with 80% to 90% involving cervical lymph nodes. Some of the common constitutional symptoms include fever, night sweats, weight loss, anemia, fatigue, and pruritus.
The tumor consists of malignant Reed-Sternberg cells and their mononuclear variants and normal reactive cells including lymphocytes and eosinophils. Classic Hodgkin’s lymphoma represents 95% of cases and has been subclassified into nodular sclerosing (60%-80%), mixed cellularity (15%-30%), lymphocyte predominant (5%), and lymphocyte depleted (<1%) variants.
Treatment consists of chemotherapy and radiation for advanced disease, with an overall 15-year survival rate of 68%. The majority of relapses occur within 3 years of treatment.
Non-Hodgkin’s Lymphoma.
About 15% of non-Hodgkin’s lymphoma arises in the head and neck, tends to occur in children between the ages of 7 and 11 years, and may involve any structure in the head and neck. There is a 3 : 1 male predominance, and patients may have widespread disease with bone involvement at diagnosis.
Clinically it presents as a neck mass or masses and can have various clinical presentations depending on the location. For example, extranodal lymphatic disease may occur in the Waldeyer’s ring, leading to middle ear effusion or nasal obstruction, or in cases of Burkitt’s lymphoma (extranodal extralymphatic) can present as a large mass involving the jaw, thyroid, or parotid gland.
The most common histologic types of non-Hodgkin’s lymphoma are malignant lymphoma, lymphoblastic (30%); malignant lymphoma, undifferentiated (Burkitt’s and non-Burkitt’s) (40%-50%); and large cell (15%-20%). Leukemia has primarily bone marrow involvement, and non-Hodgkin’s lymphoma has primarily lymph node involvement. If greater than 25% of bone marrow cells are lymphoblasts, the diagnosis is leukemia. Compared to Hodgkin’s lymphoma, non-Hodgkin’s lymphoma follows a more unpredictable course with chemotherapy and radiation therapy treatments. Prognosis is related to initial tumor stage and response to therapy.
Imaging Findings.
Imaging is often not helpful in differentiating Hodgkin’s from non-Hodgkin’s disease or metastatic lymphadenopathy. Tumor deposition sites are subdivided into nodal (most common), extranodal lymphatic (Waldeyer’s ring), and extranodal extralymphatic (orbit, paranasal sinuses, deep fascial spaces, mandible, salivary glands, skin, and larynx).
Hodgkin’s disease typically spreads from one nodal group to the next contiguous nodal group, with extranodal disease being rare ( Fig. 26-15 ). Usual presentation of this disease in children involves cervical and mediastinal adenopathy in a young patient. In non-Hodgkin’s disease, Waldeyer’s ring, paranasal sinuses, and nasal cavity are the most common extranodal sites of disease. Both can present as painless neck masses with unilateral or bilateral cervical adenopathy, with involvement of middle and lower jugular nodal chains and relative sparing of upper jugular chains.

The nodal and extranodal masses are of intermediate attenuation on CT, but on MRI demonstrate homogeneous intermediate-signal-intensity masses on all pulse sequences. On sonography, cervical adenopathy demonstrates multiple areas of decreased echogenicity, simulating cystic masses.
Evaluation of patients with head and neck lymphoma should include CT of the chest and abdomen in addition to imaging the neck. For both Hodgkin’s and non-Hodgkin’s lymphoma, gallium-67 scanning has been used for initial staging of tumor, evaluation of response to treatment, and detection of recurrent and metastatic disease. Fluorodeoxyglucose positron emission tomography (FDG PET) is increasingly becoming the imaging modality of choice.
Rhabdomyosarcoma
Soft tissue sarcomas account for 7% of all malignancies in children younger than age 15, and greater than 50% of these tumors are rhabdomyosarcomas (RMS). It is primarily a tumor of children and young adults, with most cases arising in patients younger than 12 years and 43% in patients younger than age 5. Approximately 38% are found in the head and neck. RMS is a malignant tumor that arises from primitive pluripotential mesenchymal cells capable of differentiating into skeletal muscle, and as a result these tumors may develop anywhere in the body. The most common locations for primary head and neck RMS in children are the orbit and nasopharynx, followed by paranasal sinuses and middle ear. There is a bimodal age distribution occurring at 2 to 5 years of age and the late teenage years.
Different types include embryonal, alveolar, pleomorphic, botryoid, and mixed types. More than half of RMS are embryonal variety, 70% to 90% arise in the head and neck or genitourinary tract, and most are seen in patients younger than 10 years. The Intergroup Rhabdomyosarcoma Study divided head and neck RMS into three categories based on site of origin: orbital, parameningeal, (nasopharynx, sinonasal, middle ear, mastoid, pterygoid fossa), and all other head and neck sites. The paramedian tumors extend through the skull base and invade the meninges. Tumors spread to lymph nodes of the neck in 12% to 50% of cases, and hematogenous spread also occurs to lung, bone, brain, bone marrow, and breast.
Presentation.
RMS is a solitary rapidly enlarging firm mass that involves striated muscle. Clinical presentation depends on the anatomic site of the tumor. Tumors in the neck present with a painless enlarging mass, and parameningeal RMS presents with a bloody discharge from the nose or ear, chronic otitis media with or without facial nerve palsy, or symptoms attributed to an upper respiratory infection. Tumor location also makes a difference in prognosis. For example, tumor in the orbit tends to be more localized and has a lower incidence of lymph node metastasis than other supraclavicular tumors.
Imaging Findings.
RMS is seen as a densely cellular-infiltrating neoplasm on imaging, and skeletal muscle is always seen as the site of origin. On both CT and MRI, the masses are usually homogeneous, with destruction of adjacent bone ( Fig. 26-16 ). On MRI, the tumor is isointense to muscle on T1-weighted images and hyperintense on T2-weighted images. The degree of enhancement is similar to that of normal skeletal muscle, and just over half of cases enhance homogeneously.