images with optimal renal parenchymal enhancement as well as excreted contrast material in the urinary tract. The recent development of iterative reconstruction techniques has the potential to allow for reduced dose CT imaging of the kidneys and urinary tracts with preserved image quality.6
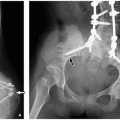
assess the male urethra in the setting of trauma (e.g., pelvic or perineal injury with hematuria) or suspected stricture.
TABLE 17.1 Routine Pediatric Voiding Cystourethrogram Images Acquired in Children with Typical Renal and Urinary Tract Anatomya | ||
|---|---|---|
|
on longitudinal ultrasound images early in life (Fig. 17.2).15 Contralateral renal parenchymal hypertrophy is often present, even early in life. CT and MRI demonstrate similar findings and can be used to search for residual renal tissue, if clinically necessary. There is no specific medical or surgical therapy for renal agenesis, and prognosis is generally good assuming the contralateral kidney is normal. Care should be taken to preserve contralateral kidney function.
 FIGURE 17.1 Anatomy of the male urethra. |
 FIGURE 17.2 1-day-old girl with right renal agenesis. No kidney is identified in the right renal fossa, and the right adrenal gland (arrows) appears abnormally elongated. |
ureteric insertion, ureterocele, or severe upper urinary tract narrowing) with resultant disordered assembly of renal tissue elements. While sometimes isolated, renal dysplasia can be associated with a variety of predisposing conditions, including upper urinary tract duplication (upper moiety parenchyma is most often dysplastic), posterior urethral valves, prune-belly syndrome, and mullerian anomalies.16,17 Microscopically, immature renal tubules embedded in primitive stroma, islands of cartilage, epithelium-lined cysts, and large aberrant blood vessels may be seen (Fig. 17.4).
 FIGURE 17.3 7-year-old boy with hypoplastic (vs. mildly dysplastic) left kidney due to congenital vesicoureteral reflux. Axial T2-weighted fat-saturated MR image shows a small but otherwise morphologically normal left kidney (arrows). There is slightly diminished left kidney corticomedullary differentiation and mild pelvicaliectasis. Right kidney is enlarged due to compensatory hypertrophy. |
 FIGURE 17.4 Hypoplastic/dysplastic kidney resected from a 9-year-old boy with a history of prune-belly syndrome (left). Microscopically (right), it showed disorganized renal elements characteristic of renal dysplasia, including nodules of abortive tubular structures surrounded by collarettes of stroma, large aberrant blood vessels, and islands of cartilage (arrow) ( hematoxylin and eosin, original magnification, 200×). |
of malignancy.20,21 In the setting of unilateral renal dysplasia, care should be taken to preserve contralateral kidney function.
 FIGURE 17.5 6-month-old girl with left kidney dysplasia. Coronal T2-weighted fat-saturated MR image shows a poorly formed left kidney with absent corticomedullary differentiation and scattered tiny parenchymal cysts (white arrows). The left ureter is obstructed due to an ectopic insertion (black arrow) and is markedly dilated. |
 FIGURE 17.6 2-month-old girl with right multicystic dysplastic kidney. A: Transverse gray-scale ultrasound image shows multiple variably sized, noncommunicating cysts (arrows) in the right renal fossa. Residual right kidney parenchyma is abnormally echogenic. B: Sagittal T2-weighted fat-saturated MR image shows similar findings (arrows). |
identified by CT and MRI. Suspicion should be high when imaging reveals an empty renal fossa. Ectopic kidneys are commonly nonrotated or malrotated, and they may have mild pelvicaliectasis, even in the absence of urinary tract obstruction or VUR (Fig. 17.8). The arterial supply and venous drainage of ectopic kidneys are usually anomalous, with renal arteries arising from nearby major arterial structures and renal veins draining to nearby major systemic venous structures. Ectopic kidneys are often asymptomatic but may be complicated by ipsilateral VUR, UPJ obstruction, and urolithiasis.25 Small, poorly functioning malpositioned kidneys rarely can present with urinary dribbling and/or incontinence due to an associated ectopic ureter.26
 FIGURE 17.7 3-year-old girl with nonrotated right kidney. Axial postcontrast T1-weighted fat-saturated MR image shows nonrotation of the right kidney, with an anteriorly oriented renal pelvis (arrow). The left renal fossa is empty. |
 FIGURE 17.8 11-year-old boy with pelvic kidney. Sagittal contrast-enhanced CT image shows that the left kidney (arrows) is located in the pelvis, posterior to the bladder (B). The kidney is nonrotated with its mildly dilated renal collecting system (asterisk) facing anteriorly. |
kidneys may be associated with VUR, UPJ obstruction, and urolithiasis. Cross-fused renal ectopia also generally requires no specific treatment.
 FIGURE 17.9 3-month-old boy with horseshoe kidney. Transverse gray-scale ultrasound image through the midabdomen shows fusion of the bilateral lower renal poles in the midline (arrow), anterior to the lumbar spine. |
 FIGURE 17.10 7-year-old boy with horseshoe kidney. A: Axial T2-weighted fat-saturated MR image shows abnormal rotation of the kidneys and mild left pelvicaliectasis (asterisk). B: More inferior MR image shows that the kidneys are fused in the midline with a thick parenchymal isthmus (arrow). (Case courtesy of J. Damien Grattan-Smith, MD, Children’s Healthcare of Atlanta, Atlanta, GA.) |
 FIGURE 17.11 13-year-old boy with injured horseshoe kidney due to motor vehicle accident. Axial contrast-enhanced CT image shows a lacerated horseshoe kidney with adjacent retroperitoneal hematoma (between arrows). Renal collecting system disruption was identified on delayed excretory-phase imaging. |
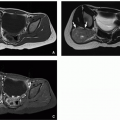
often associated with infection by Proteus mirabilis, a ureasplitting bacterium that forms struvite calculi. At histology, XGP is characterized by the presence of chronic inflammation, including lipid-laden macrophages. At imaging, a large obstructing calculus, sometimes staghorn in appearance, classically is present in the renal pelvis. The affected kidney usually appears diffusely enlarged and contains numerous round areas of decreased echogenicity at ultrasound or decreased attenuation at CT that are due to severe hydronephrosis (“bear paw” sign) and parenchymal necrosis (Fig. 17.15). Extensive perinephric inflammatory changes are typically present, and abscess formation may occur in the perinephric space, adjacent psoas muscle, and even body wall. Delayed contrast-enhanced CT and renal scintigraphy reveal minimal or no renal function, and radical nephrectomy is generally indicated for definitive treatment. A minority of XGP cases are focal, involving only a portion of the kidney, sometimes appearing mass-like.36
 FIGURE 17.12 3-month-old boy with crossed fused renal ectopia. A: Voiding cystourethrogram image shows vesicoureteral reflux into two separate renal collecting systems (arrows), both located to the left of midline. B: Gray-scale ultrasound image confirms that the right kidney is ectopic and fused to the left kidney. A parenchymal notch (arrow) can be seen at the site of parenchymal fusion. |
 FIGURE 17.13 13-year-old boy with fever, right flank pain, and hematuria due to pyelonephritis. A: Longitudinal color Doppler ultrasound image of the right kidney shows a mass-like area of increased echogenicity (arrows) in the upper pole that has decreased blood flow compared to adjacent parenchyma. B: Axial contrast-enhanced CT image shows an ill-defined low attenuation area (arrows) in the right kidney upper pole due to parenchymal infection. This area of acute focal bacterial pyelonephritis resolved on follow-up ultrasound imaging after antibiotic therapy. |
Ascending fungal infection within the renal collecting system is best appreciated by ultrasound, sometimes presenting as nonspecific echogenic debris in the urine or as an echogenic, circumscribed, sometimes mobile fungus ball (mycetoma) (Fig. 17.16).38 On occasion, ascending infection can infiltrate the renal medulla. Renal or perinephric abscesses due to fungal infection can rarely occur. Treatment is typically antifungal medical therapy, although, in certain children, asymptomatic funguria is due to colonization and does not require medical therapy. Rarely, such fungal infections can result in urinary tract obstruction or nonresolving abscesses that require percutaneous drainage or surgical management.39
 FIGURE 17.14 13-year-old girl with fever and right flank pain. Axial contrast-enhanced CT image shows a heterogeneous abscess (arrow) in the anterior aspect of the mid right kidney. Perinephric fluid surrounds much of the right kidney. |
 FIGURE 17.15 1-year-old girl with recurrent fevers due to xanthogranulomatous pyelonephritis. A: Axial contrast-enhanced CT image shows marked enlargement of the right kidney. Multiple calculi (white arrow) are present in the right renal collecting system. Numerous areas of focal low attenuation in the right kidney are due to pelvicaliectasis and parenchymal necrosis. A right retroperitoneal abscess (black arrow) is present adjacent to the right psoas muscle. Enlarged retroperitoneal lymph nodes are reactive in etiology. B: Bivalved gross pathologic specimen shows extensive renal parenchymal necrosis. |
 FIGURE 17.16 4-month-old girl with fungemia and funguria. Longitudinal gray-scale ultrasound image of the right kidney shows an echogenic, lobulated fungus ball (arrows) in upper pole collecting system. |
 FIGURE 17.17 Congenital mesoblastic nephroma, resected from a 2-month-old girl. The 9 cm mass expanded and occupied the majority of the kidney tissue, showing a poorly demarcated border with normal kidney (left). Microscopically, the tumor consisted of a cellular fibroblastic proliferation, showing intermixed residual nonneoplastic kidney including the benign medullary tubules depicted here (right, hematoxylin and eosin, original magnification, 200×). This tumor harbored an ETV6 gene rearrangement typical of cellular congenital mesoblastic nephroma, and the patient had a high serum calcium level attributed to paraneoplastic disease. |
 FIGURE 17.18 7-month-old girl with right congenital mesoblastic nephroma (cellular variant). Axial contrast-enhanced CT image demonstrates a very large, heterogeneous mass (arrows) arising from the right kidney with substantial mass effect upon adjacent structures. |
 FIGURE 17.19 10-month-old girl with ossifying renal tumor of infancy. Axial contrast-enhanced CT image shows a partially calcified mass (arrows) centered in the left renal collecting system. Left caliectasis and renal parenchymal thinning are due to chronic obstruction. (Image courtesy of Edward Y. Lee, MD, MPH, Boston Children’s Hospital and Harvard Medical School, Boston, MA.) |
 FIGURE 17.20 Cystic nephroma in a 13-year-old girl with a known DICER1 gene mutation and previous cervicovaginal rhabdomyosarcoma. The cut surface (left) shows a well-delineated 4.5 cm mass containing multiple cysts filled with clear fluid. Microscopically, the lesion shows epithelium-lined cysts with condensation of stromal cells immediately underlying the epithelium (right, hematoxylin and eosin, original magnification, 100×). |
 FIGURE 17.21 Cystic partially-differentiated nephroblastoma in an 11-month-old girl with a palpable abdominal mass. Axial contrast-enhanced CT image shows a large, cystic, heavily septated, centrally located mass (arrows) within the right kidney. |
hemihypertrophy, macrosomia, midline abdominal wall defects, ear anomalies, and/or neonatal hypoglycemia), sporadic aniridia, WAGR syndrome (Wilms tumor, Aniridia, Genitourinary anomalies, and mental Retardation), and Denys-Drash syndrome (gonadal dysgenesis, mesangial renal sclerosis leading to nephrotic syndrome and chronic kidney disease). All of these predisposing conditions relate to abnormalities of chromosome 11 and the WT1 and WT2 genes.54,55
 FIGURE 17.22 7-year-old girl with tuberous sclerosis, numerous bilateral renal angiomyolipomas (AMLs), and left renal cell carcinoma. A: Longitudinal gray-scale ultrasound image of the right kidney shows many small echogenic parenchymal lesions, consistent with AMLs. B: Longitudinal gray-scale ultrasound image through the left kidney shows multiple punctate echogenic AMLs as well as a 3.5 cm dominant echogenic mass (arrows) in the upper pole. Image-guided core needle biopsy of the large upper pole lesion revealed renal cell carcinoma, which was confirmed at surgical pathology. |
and moderate specificity for identifying preoperative Wilms tumor rupture, with extension of ascites beyond the pelvic cul-de-sac being the best indicator.58
 FIGURE 17.23 13-year-old girl with tuberous sclerosis and numerous bilateral renal angiomyolipomas. Axial out-of-phase T1-weighted gradient-recalled echo MR image demonstrates numerous areas of signal loss (arrows) in the kidneys (“India ink” artifact) due to the presence of lipid and water in the same voxel. |
 FIGURE 17.24 17-year-old girl with life-threatening left retroperitoneal hemorrhage. Multiple bilateral low-attenuation and enhancing renal lesions representing a combination of cysts and angiomyolipomas. High-attenuation perinephric hematoma (arrows) is due to a bleeding angiomyolipoma. |
 FIGURE 17.25 3-year-old boy with bilateral Wilms tumors. The left image shows triphasic differentiation, including blastemal, stromal, and epithelial elements, which here are forming abortive tubules and glomeruli (hematoxylin and eosin, original magnification, 200×). These tumors arose in a background of nephroblastomatosis (right), illustrated here by a perilobar nephrogenic rest (hematoxylin and eosin, original magnification, 20×). |
 FIGURE 17.26 14-month-old boy with a palpable abdominal mass due to left Wilms tumor. A: Axial contrast-enhanced CT image shows a large, heterogeneously enhancing mass arising from the left kidney extending into the left renal vein (arrows). The “claw sign” is present. B: Coronal contrast-enhanced CT image shows that the mass extends into the inferior vena cava (white arrow). There are multiple pulmonary metastases (black arrows). |
rind-like soft tissue abnormality replacing the renal cortex (Fig. 17.28).61,62
 FIGURE 17.27 11-month-old girl with Beckwith-Wiedemann syndrome, bilateral nephroblastomatosis, and presumed bilateral Wilms tumor. Coronal contrast-enhanced CT image shows multiple bilateral low-attenuation renal masses (asterisks) due to nephrogenic rests and multifocal Wilms tumor. |
 FIGURE 17.28 2-week-old boy with multiple congenital anomalies. Axial contrast-enhanced CT image shows right nephromegaly and rind-like parenchymal thickening (arrows) due to diffuse nephroblastomatosis. The patient was later treated with chemotherapy for presumed Wilms tumor as this abnormality enlarged during infancy. |
 FIGURE 17.29 11-year-old girl with left flank pain due to renal cell carcinoma. Axial contrast-enhanced CT image shows a heterogeneously enhancing mass (arrows) arising from the left kidney. A large amount of high attenuation fluid (asterisks) in the left perinephric space is due to hemorrhage. Mildly enlarged retroperitoneal lymph nodes were proven to contain metastatic neoplasm. |
 FIGURE 17.30 1-year-old boy with palpable abdominal mass due to clear cell sarcoma of the kidney. Axial contrast-enhanced CT image shows a large, heterogeneous mass (arrows) arising from the left kidney. The “claw sign” is present. |
 FIGURE 17.31 1-year-old girl with rhabdoid tumor of the kidney. Coronal contrast-enhanced CT image shows a heterogeneously enhancing mass (arrows) in the upper pole of the left kidney. There is a large subcapsular fluid collection (asterisks) that is causing mass effect on normal left kidney parenchyma. (Image courtesy of Edward Y. Lee, MD, MPH, Boston Children’s Hospital and Harvard Medical School, Boston, MA.) |
lymphatic invasion. Renal parenchymal satellite lesions are also common.73 This neoplasm has an extremely poor prognosis with survival typically <6 months, and it is frequently metastatic at the time of diagnosis.74
 FIGURE 17.32 21-year-old young man with sickle cell trait and right kidney medullary carcinoma. Axial contrast-enhanced CT image shows a large, heterogeneous, infiltrative mass (arrows) located centrally in the right kidney. Coronal CT images (not shown) revealed extension of tumor into the proximal right ureter. |
 FIGURE 17.33 14-year-old girl with flank pain and hematuria due to right kidney Ewing sarcoma/primitive neuroectodermal tumor (PNET). Axial contrast-enhanced CT image shows a large, enhancing mass arising from the right kidney. The mass extends into the right renal vein and inferior vena cava (arrows). |
include a solitary renal mass or large retroperitoneal mass engulfing the kidney.80 Nearby retroperitoneal lymph node enlargement is common. Renal lymphomatous deposits typically regress with chemotherapy.
 FIGURE 17.34 5-year-old boy with renal involvement by Burkitt lymphoma, identified after presentation with a palatine tonsillar mass. Coronal contrast-enhanced CT image shows bilateral hypoenhancing solid renal masses (asterisks) due to lymphomatous deposits. |
 FIGURE 17.35 12-year-old boy with acute leukemia. Axial contrast-enhanced CT image shows very large areas of geographic low attenuation in both kidneys due to leukemia. The kidneys are enlarged, and abnormal soft tissue (arrows) encases the abdominal aorta. |
TABLE 17.2 American Association for the Surgery of Trauma Kidney Injury Grading Scale | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
vary in size from very small to quite large.85,86 Extravasation of intravascular contrast material on contrast-enhanced CT suggests active bleeding that may require treatment (e.g., arterial embolization). On occasion, hemorrhage may be confined between the renal capsule and underlying parenchyma. Subcapsular hematomas are generally crescentic or elliptical in shape and exert mass effect upon underlying renal parenchyma (Fig. 17.38). Acute and subacute subcapsular hematomas demonstrate higher attenuation than water at CT. On occasion, subcapsular hematomas and associated renal compression may cause renal failure and renin-mediated hypertension (so-called Page kidney).87
 FIGURE 17.36 12-year-old boy in bicycle accident with right kidney laceration. A: Axial contrast-enhanced nephrographic phase CT image shows a large area of posterior right kidney hypoenhancement (between arrows). A large amount of fluid (blood and/or urine) is present in the right retroperitoneum. B: Axial contrast-enhanced excretory-phase CT image shows extravasation of excreted contrast material (arrows) into the right perinephric space due to collecting system injury. |
 FIGURE 17.37 13-year-old boy with blunt abdominal trauma and left renal artery injury. Axial contrast-enhanced excretory-phase CT image shows complete absence of left kidney parenchymal enhancement (asterisks). A contrast-filled pseudoaneurysm (arrow) is present in the left renal hilar region. |
blood flow as well as “arterialization” of venous blood flow with increased turbulence (Fig. 17.39).85 Many of these lesions are small and involute without intervention, although large lesions may persist and require arterial embolization.86
 FIGURE 17.38 8-year-old boy with obstructed right kidney due to large intra-abdominal rhabdomyosarcoma. Axial contrast-enhanced CT image shows a traumatic right kidney subcapsular hematoma (arrow) related to percutaneous nephrostomy tube placement. The hematoma has mass effect upon adjacent renal parenchyma. |
 FIGURE 17.39 11-year-old boy with left lower quadrant renal allograft status post percutaneous biopsy. A: Longitudinal spectral Doppler ultrasound image shows a focus of very high arterial peak systolic velocity with increased diastolic blood flow in the lower pole of the transplant kidney (arrow), consistent with arteriovenous fistula from recent biopsy. B: Interrogation of an interpolar arcuate artery shows a normal spectral Doppler waveform. |
often diagnosed during adulthood, it can present during childhood. ADPKD is classified as a “ciliopathy” and can be due to a mutation in either the PKD1 (85%) or PKD2 (15%) gene.91
 FIGURE 17.40 16-year-old girl with incidentally detected simple renal cyst. Coronal postcontrast T1-weighted fat-saturated MR image shows a 1.5-cm circumscribed, nonen-hancing lesion (arrow) in the right kidney upper pole. There is no internal complexity. |
 FIGURE 17.41 3-year-old girl with palpable abdominal mass due to a large left renal simple cyst. Axial T2-weighted fat-saturated MR image demonstrates a large left kidney cyst (arrows) without mural thickening or internal complexity. |
 FIGURE 17.42 9-year-old girl with enuresis. Longitudinal gray-scale ultrasound image through the left kidney lower pole shows a complicated cyst (arrows) containing multiple septations. |
 FIGURE 17.43 7-year-old boy with autosomal dominant polycystic kidney disease. Coronal contrast-enhanced CT image shows multiple bilateral simple renal cysts. The left kidney is enlarged, and a very large cyst (arrows) arises from its upper pole. |
have less severe renal involvement. Such abnormalities include congenital hepatic fibrosis and bile duct ectasia (Caroli syndrome), with associated splenomegaly, portosystemic varices, and ascites due to portal hypertension.96 Almost all individuals affected by ARPKD require dialysis or renal transplantation by adulthood, sometimes very early in life.
 FIGURE 17.44 Diffusely enlarged (14.5 cm) kidney from a 6-day-old girl with autosomal recessive polycystic kidney disease who underwent bilateral nephrectomy to ameliorate respiratory distress. Close gross inspection (right) shows innumerable small cysts, some oriented with their long axis perpendicular to the kidney capsule. |
 FIGURE 17.45 6-month-old boy who presented with cardiac arrest, respiratory failure, and marked abdominal distention. Longitudinal gray-scale ultrasound image through the left kidney reveals nephromegaly and loss of parenchymal corticomedullary differentiation. Numerous tiny cystic structures are present throughout the kidney. Findings are consistent with autosomal recessive polycystic kidney disease. The left renal collecting system is mildly dilated (asterisks). |
 FIGURE 17.46 19-year-old man with von Hippel-Lindau disease. Axial T2-weighted fat-saturated MR image shows a centrally located simple cyst in the left kidney (white arrow; two other small cysts were also present in the left kidney, not shown). Multiple small cystic lesions (black arrow) are also present in the pancreas. |
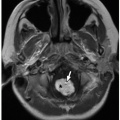
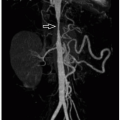
measure pressure gradients, as well as evaluate the aorta (Figs. 17.48 and 17.49). CBDSA has superior spatial resolution compared to other imaging modalities, detects accessory renal artery stenoses, and allows for simultaneous endovascular therapy of certain lesions.102 Renal artery stenoses are frequently intrarenal in children,102 and bilateral lesions occur in about 40%.103 Aneurysm formation is also common, often poststenotic in location (Fig. 17.49).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


