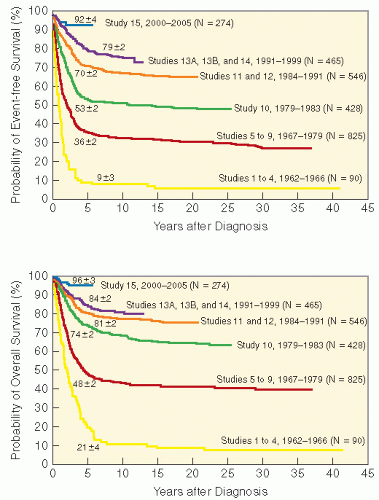
 Figure 2.1 Outcome in childhood ALL—serial “total” therapy studies at St. Jude Children’s Research Hospital 1962-2005. EFS (top) and OS (bottom) for 2628 children treated on 15 consecutive trials. Reprinted from Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166-178, with permission. |
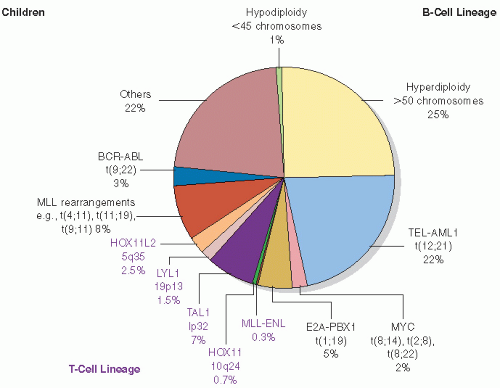 Figure 2.2 Genotypes in childhood ALL, including genetic findings in B-cell lineage or all ALL immunophenotypes; genetic findings unique to T-cell ALL are in purple shades. Reprinted from Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15): 1535-1548, with permission. |
t(9;22)/BRC-ABL fusion (Ph+ ALL) (14,15). In addition to clinical and biological features at diagnosis, one of the most important predictors of outcome is early response to induction chemotherapy: patients with residual bone marrow leukemia (MRD quantitatively defined by immunohistochemistry or polymerase chain reaction [PCR] blasts defined as ≥1% on day 19 of induction therapy or 0.1-0.99% at completion of 6-week induction therapy) are at higher risk for relapse and are staged as standard risk (rather than low risk); those with ≥1% residual at the latter interval are all treated as high risk (14,16). Relative risk factors and the impact on therapeutic outcome are listed in Table 2.1. Recent studies show 45-50% of cases treated as low risk, 40-45% as standard risk, and 8-10% as high risk (14,17). With more aggressive, risk-adapted therapy, many of the outcome correlates are no longer significant, including mature B-cell or T-cell immunophenotypes, race, and gender (5,14,18).
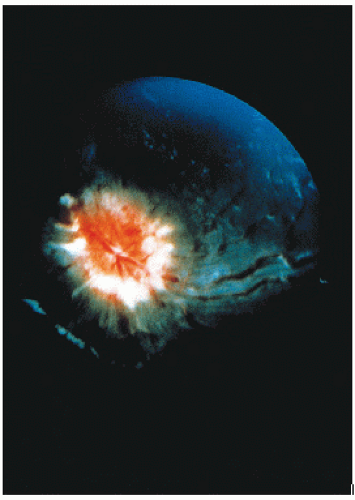 Figure 2.3 The retinal photograph demonstrates advanced CNS leukemia with papilledema and retinal infiltrate typical of ocular disease. |
relapse from >60% to <5-10% (32,35). Children in hematologic remission randomized to preventive CSI (24 Gy) or equivalent therapeutic CSI at the time of overt CNS relapse showed strikingly higher event-free and overall survival with preventive therapy (36). In addition, children cured following a CNS relapse had significantly greater functional deficits (e.g., seizure disorders and cognitive deficits) (37). Subsequent trials proved the relative equivalence of 18 Gy (at 1.5-1.8 Gy per fraction) and 24 Gy (similarly fractionated) for preventive CrI (38, 39, 40, 41).
Table 2.1 Preventive CNS Therapy in Acute Lymphoblastic Leukemia—Major Recent Series | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
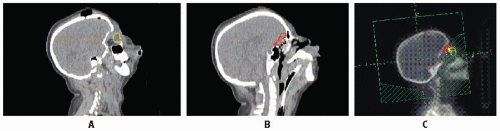 Figure 2.4 Craniospinal irradiation for CNS leukemia. A: Sagittal CT simulation reconstruction showing eye (yellow) and optic nerve (blue) at midorbital level; cranial irradiation for CNS leukemia includes the subarachnoid extension along the optic nerve sheath and the posterior retina (see Fig. 2.7). B: Midline sagittal CT simulation reconstruction showing the cribriform plate (red) along the lower midline skull base anteriorly; this is a critical target for full cranial irradiation. C: Outline of cranial irradiation volume providing margin at key anatomic regions (A and B) for craniospinal therapy in ALL. |
further CrI dosage reduction (12 Gy/8 fractions in the more recent BFM studies) (39).
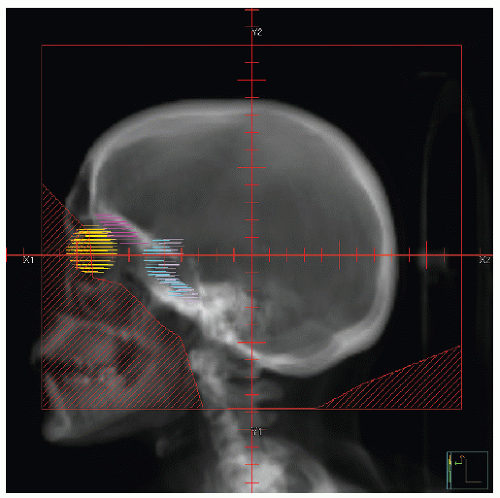 Figure 2.5 Lateral field for cranial irradiation in ALL. Note margin below cribriform plate (pink) and middle cranial fossa (blue). Targeted volume includes posterior eye (yellow) and orbit. |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


