© Springer International Publishing AG 2017
Pietro Ruggieri, Andrea Angelini, Daniel Vanel and Piero Picci (eds.)Tumors of the Sacrum10.1007/978-3-319-51202-0_1818. Lymphoma and Myeloma of the Sacrum
Andreas F. Mavrogenis1 , Georgios N. Panagopoulos1, Andrea Angelini2 , Pier Luigi Zinzani3 and Pietro Ruggieri2
(1)
First Department of Orthopaedics, National and Kapodistrian University of Athens, Athens, Greece
(2)
Department of Orthopedics and Orthopedic Oncology, University of Padova, Padova, Italy
(3)
Institute of Hematology “L. E A. Seràgnoli”, University of Bologna, Bologna, Italy
18.1 Introduction
Neoplasms of the sacrum are relatively uncommon, representing 1–2% of all musculoskeletal tumors [1]. They can be classified as primary or metastatic, the former being either benign or malignant. The most common malignancy of the sacrum is metastatic disease, whereas the most common primary sacral tumor is chordoma; more rare sacral malignancies include hematologic neoplasms, Ewing’s sarcoma, chondrosarcoma, and osteosarcoma [2].
Hematologic malignancies, more frequently multiple myeloma and lymphoma, occur relatively frequently in the sacrum, accounting for 18% of all primary malignant sacral tumors [3]. Traditional treatment for hematologic neoplasms affecting bone consists of chemotherapy and/or radiation therapy [4]. As medical treatment of these conditions improves and patients’ survival extends, skeletal lesions caused by hematologic malignancies are more likely to necessitate some form of orthopaedic surgery or interventional procedure, with either curative or palliative intent.
18.2 Lymphoma
Primary lymphoma of bone is rare, accounting for 3–7% of bone tumors and less than 2% of adult lymphomas [5, 6]. Although there is no universal definition, by convention, an interval of 4–6 months between skeletal manifestation of the lesion and the development of extraskeletal disease is required for the tumor to be considered a primary tumor in bone [5]. Because of the infrequency of primary bone lymphoma and the disagreement as to its clinical definition, there have been few published series documenting patients’ outcomes, clinical characteristics, and biological aspects [5–17].
The great majority of bone lymphomas are diffuse large B-cell Non-Hodgkin lymphomas. In three published large series of primary bone lymphoma, this histotype was represented in at least 70, 79, and 83% of cases [7–9]. More than 50% of primary lymphomas of bone occur in patients older than 60 years, with a slight male predominance [8]. The vertebrae, femur, and pelvis are the most frequently involved bones, accounting for approximately 29%, 12%, and 13% of lesions, respectively. Lesion multifocality is seen in approximately 15% of cases [8].
The incidence of primary sacral lymphoma is unknown, due to its rarity, with only a few case reports dedicated to the subject [2, 10]. Similarly to other sacral lesions, persistent low back pain from a sacral insufficiency fracture, and referred pain to a leg or buttock from nerve root irritation, are the usual presenting symptoms [2]. Neurological signs are rarely seen prior to diagnosis or tend to manifest late in the course of disease. If the lesion has expanded into the presacral space, rectal exam may reveal the presence of a palpable soft tissue mass.
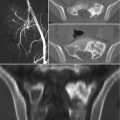
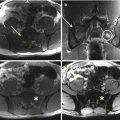
Radiographic features are usually non-specific and tend to overlap with those of other tumors. As a rule of thumb, lymphoma of bone will present as a lytic destructive lesion with a permeative or moth-eaten pattern [5]. CT and MR imaging are helpful in evaluating the extent of bone involvement and cortical erosion, as well as the soft tissue extension of the tumor. Bone scan or PET/CT are useful to document additional foci of bone or extraskeletal involvement [11].
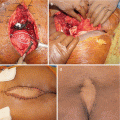
The Ann Arbor staging system developed for Hodgkin’s disease has also been used in staging non-Hodgkin lymphomas [12]. This staging system focuses on the number of tumor sites (nodal and extranodal), location, and presence/absence of systemic symptoms [12]. Percutaneous biopsy of sacral lesions is a reasonable first-line diagnostic tool. Fine-needle aspiration biopsy sampling performed using CT, fluoroscopic, or ultrasound guidance has been reported to be successful [13]; although the use of fine-needle biopsy in musculoskeletal tumors is not recommended. Because there is a 7% rate of false-negative findings, a negative biopsy result should prompt a repeated biopsy procedure, preferably using a core or trephine tool, or an open biopsy [2].
Immunohistochemistry is usually essential for diagnosis. Lymphoma must be distinguished from other hematopoietic neoplasms, such as plasma cell myeloma, Langerhans cell histiocytosis, and mastocytosis. The differential diagnosis should also include round-cell tumors, such as Ewing’s sarcoma, rhabdomyosarcoma, and metastatic small cell carcinoma. Most untreated diffuse large B-cell lymphomas are CD20-positive. If this is negative, PAX5 or CD79 will usually be positive. Once the diagnosis is confirmed, further prognostic tests, such as Ki-67, CD10, BCL6, and MUM1, are usually requested to subclassify the disease and guide treatment [14]. Radiation therapy and chemotherapy are the standard treatments for primary lymphoma of bone. A general algorithm for stage I/II, nonbulky disease involves 3–4 cycles of rituximab plus cyclophosphamide, vincristine, doxorubicin, and prednisone (CHOP), followed by involved field radiation therapy (IFRT) [15–17].
18.3 Multiple Myeloma
Multiple myeloma is a monoclonal, neoplastic proliferation of plasma cells that involves the bone marrow and occasionally extraskeletal sites, whose prominent feature is the widespread presence of lytic bone lesions [14]. It is one of the most frequently occurring hemapoietic malignancies (10%), accounting for approximately 1% of all malignant neoplasms in White and 2% in Black patients [14]. It is also the most frequent neoplasm to present with skeletal lesions, with the skull, vertebral bodies, pelvis, and proximal parts of long bones being the most commonly involved sites [18, 19]. Peak incidence is between ages 65 and 74 years, with a median age at diagnosis ranging from 66 to 69 years [20, 21].
It is currently accepted that myeloma is always preceded by (or evolves from) a premalignant condition clinically recognized as monoclonal gammopathy of undetermined significance (MGUS) [22]. MGUS is usually asymptomatic and present in 3–4% of the general population older than 50 years. It is associated with a risk of progression to multiple myeloma of approximately 1% per year [23]. Smoldering multiple myeloma (SMM) is an intermediate stage between MGUS and multiple myeloma and is associated with a higher risk of progression of approximately 10% per year [24].
The most common presenting symptoms of multiple myeloma are fatigue and bone pain [25]. Anemia is detected in 75% of patients, whereas osteolytic skeletal lesions are present in 80% of cases [21]. Other findings may include hypercalcemia, renal failure, proteinuria, and a history of recurrent infections [14]. The cardinal diagnostic feature of multiple myeloma is the detection of monoclonal (M) protein on serum or urine protein electrophoresis (82% of patients) [25]. Serum immunofixation, serum-free light chain (FLC) assay, or 24 h urine studies can further increase sensitivity of detection [26]. The M protein type is IgG in approximately 50%, IgA in 20%, immunoglobulin light chain only in 20%, IgD in 2%, and IgM in 0.5% [25]. About 2–3% of multiple myeloma has no detectable M protein and is referred to as nonsecretory multiple myeloma [27].
The baseline diagnostic work-up required for the diagnosis of multiple myeloma includes a complete blood cell count, measurement of serum calcium and creatinine levels, serum and urinary protein electrophoresis with immunofixation, serum FLC assay, and bone marrow examination [21]. Plain radiography of the entire skeleton (skeletal survey) to detect osteolytic bone lesions usually completes the diagnostic work-up [28]. From a biopsy standpoint, diagnosis of multiple myeloma requires the presence of clonal bone marrow plasma cells of ≥10%, or biopsy-proven bony or extramedullary plasmacytoma [29]. Skeletal lesions typically represent multifocal, sharply demarcated, lytic, often called punched-out lesions. CT and MR imaging are useful adjuncts in the spine, or when skeletal survey is initially negative.
Although multiple myeloma is still considered by many to be a single disease, it is in reality a collection of several cytogenetically distinct plasma cell neoplasms. These distinct forms of myeloma are designated as solitary plasmacytoma of bone, extramedullary plasmacytoma, nonsecretory myeloma, plasma cell leukemia, and osteosclerotic myeloma (POEMS) [14]. Amyloidosis associated with multiple myeloma can also cause bone lesions (amyloidomas) [30]. Although median survival in patients with multiple myeloma is approximately 5–7 years, there is major variation in survival depending on host factors, tumor burden (stage), biology (cytogenetic abnormalities), and response to therapy [31]. Based on these premises, risk stratification is carried out, which will subsequently lead to treatment. Molecular classification is important, as high-risk cytogenetic features are likely to modify therapy accordingly.
Until 2000, the mainstay of therapy for multiple myeloma was the use of alkylators and corticosteroids, and in selected patients, high-dose chemotherapy with autologous stem cell transplant (ASCT) [21]. Subsequently, alternative agents, such as thalidomide, bortezomib, and lenalidomide, have emerged as effective agents that have significantly improved clinical outcome [32–35]. More recently, carfilzomib, pomalidomide, panobinostat, daratumumab, ixazomib, and elotuzumab have been approved for the treatment of multiple myeloma in the United States, substantially expanding the number of treatment regimens available for patients in all stages of the disease [21]. In patients diagnosed with multiple myeloma, development of end-organ damage (myeloma-defining events) is the hallmark indication for treatment initiation [20]. Evidence of end-organ damage is traditionally represented by the acronym CRAB (hyperCalcemia, Renal insufficiency, Anemia, lytic Bone lesions) [18, 36, 37]. Multiple myeloma without end-organ damage is referred to as smoldering multiple myeloma (SMM) and should be monitored closely, as these patients have a risk of progression of approximately 10% per year for the first 5 years, 3% per year for the next 5 years, and 1% per year thereafter [24].
The most important phases of therapy are initial therapy, ASCT (if eligible), consolidation/maintenance therapy, and treatment of relapse [21]. Transplant-eligible patients typically receive approximately four cycles of initial therapy followed by stem cell collection and ASCT. Delayed ASCT, with stem cell collection after four cycles of initial therapy and cryopreservation for future use, is also possible. Pre-transplant conditioning (total body irradiation and/or melphalan) is necessary prior to ASCT [38]. ASCT has been shown to increase median survival in MM by approximately 12 months [39]. The upper limit for ASCT is 65 years in most countries (75 years in the US) [20, 21]. Transplant-ineligible patients are usually treated for 12–18 months. After initial therapy, with or without ASCT, consolidation/maintenance therapy should be considered. Treatment at relapse or progression is more complicated and may include re-induction and repeat ASCT, or treatment with novel drugs [20].
Supportive care is also paramount in patients with multiple myeloma. Hypercalcemia is usually treated with hydration, steroids, and bisphosphonates (pamidronate or zoledronic acid). Pamidronate at 60–90 mg intravenously over 2–4 h, or zoledronic acid at 4 mg intravenously over 15 min, will normalize the calcium levels within 24–72 h in most patients [40, 41]. In refractory patients, salmon calcitonin may be used. A small percentage of patients with multiple myeloma may develop hyperviscosity syndrome (especially in the IgA subtype). This should be promptly treated with plasmapheresis [42]. Prevention of recurrent infections is another important issue. Patients with MM should receive pneumococcal and influenza vaccinations. Intravenously administered gammaglobulin every 3–4 weeks is indicated if patients have recurrent serious infections associated with severe hypogammaglobulinemia [21]. Prophylaxis against herpes zoster reactivation and Pneumocystis jiroveci should also be considered [43].
Maybe the most important element in the supportive care of patients with multiple myeloma is the use of bisphosphonates to prevent and/or reduce the number of skeletal events and, ultimately, the associated pain and morbidity. Zoledronic acid or pamidronate once monthly at least for the first 1–2 years is recommended for almost all patients with multiple myeloma who have evidence of myeloma-associated bone disease [44–48]. All patients should receive a dental examination before initiation of bisphosphonates, and invasive dental procedures should be done with caution because of the risk of osteonecrosis of the jaw [49]. Local radiation therapy is also used in patients with pain refractory to analgesics and systemic therapy, as well as in case of spinal cord compression with extramedullary tumor extension. Vertebroplasty and kyphoplasty are frequently used for vertebral pathological fractures [50]. Prophylactic nailing is also commonly performed for pathological or impending fractures of long bones. Sacroplasty is also evolving as an acceptable and efficient treatment of MM-induced sacral insufficiency fractures. Even though published series are limited, results intended primarily as pain relief seem to be promising [51–53].
Treatment is different in the case of localized disease, such as solitary plasmacytoma or extraosseous plasmacytoma. These cases can be treated either with curatively intended radiation therapy to the involved field with cumulative doses of ≥45 Gy [54, 55], or resected surgically [4, 20]. Although cure is the primary goal of treatment in solitary plasmacytoma, a progression to multiple myeloma may occur in 30–60% of cases within the next 3–5 years, thereby requiring regular follow-up [56].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


