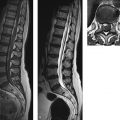
Congenital malformations of the spinal column are generally diagnosed immediately after birth or during the first years of life; rarely, they are discovered in older children or adults. The timing of the diagnosis correlates closely with the severity of the malformation. The task of MRI in this setting is twofold: (1) to define the anatomy of the malformation as accurately as possible in order to classify it and select patients for surgical treatment and (2) to diagnose postoperative complications. A precise knowledge of embryology is essential in this context in order to detect frequent combinations of anomalies occurring at noncontiguous sites or to predict future developments and surgical outcomes. Moreover, clinical manifestations such as subcutaneous swellings or skin changes can have a major bearing on diagnostic imaging, as they can critically influence the selection of neuroradiology techniques. The spinal column and spinal cord are easily accessible to ultrasound imaging during the first year of life. A variety of malformations can be detected with ultrasound, although the full extent of coexisting anomalies often cannot be appreciated. CT is also used as an adjunct in certain anomalies that involve complex osseous changes, for example, or for the classification of diastematomyelias. The imaging modality of choice for both the exclusion and diagnosis of spinal malformations is MRI. The scope of the examination should generally cover the entire spinal column to permit the detection of complex malformations and combinations of anomalies. Besides standard axial and sagittal sequences, coronal images are of key importance due to the frequency of associated vertebral anomalies. Three-dimensional T1w and T2w sequences with 1-mm slices and multiplanar reconstructions can be particularly informative in smaller children. Additional imaging in the prone position has proven useful in the diagnosis of tethered cord. The protocol should include intravenous contrast administration in cases where there is clinical suspicion of a dermal sinus or enteric fistula. Note Imaging should generally cover the entire spinal column including the craniocervical junction to permit the detection of complex or combined malformations. Spinal malformations result from disturbances of normal development during the period from the third to sixth gestational weeks. The relevant embryologic steps are gastrulation (weeks 2–3), primary neurulation (weeks 3–4), and secondary neurulation and retrogressive differentiation (weeks 5–6). “Gastrulation” is a term used in embryogenesis to denote the invagination and folding of the embryo to form the gastrula with an outer germ layer (epiblast), inner germ layer (hypoblast), and later a middle germ layer (mesoblast; ▶ Fig. 17.1, ▶ Fig. 17.2). The main purpose of gastrulation is to form the dorsal primitive streak and dorsal notochord. Fig. 17.1 Gastrulation. Diagrammatic representation. Changes in the embryonic disk in the third week of gestation (dorsal view), characterized by formation of the primitive streak and primitive node and development of the notochordal process and neural plate (from Jansen O, Stephani U. RRN Fehlbildungen und frühkindliche Schädigungen des ZNS. Stuttgart: Thieme; 2007). (a) Days 14–15. (b) Day 16. (c) Approximately day 18. Fig. 17.2 Development of the notochord. Diagrammatic representation (from Jansen O, Stephani U. RRN Fehlbildungen und frühkindliche Schädigungen des ZNS. Stuttgart: Thieme; 2007). (a) Day 16: The notochord canal develops as a prolongation of the primitive groove. (b) Day 17: The floor of the canalized notochordal canal fuses with the endoderm and degenerates, so that the amniotic cavity and yolk sac communicate via the neuroenteric canal. (c) Day 22: The definitive notochord forms from the infolded chordal plate, and the endoderm recloses over it. At the end of just the first gestational week, the blastula differentiates into two parts: the epiblast (embryonic ectoderm) and the hypoblast (embryonic endoderm). On days 14 to 15, a cord of thickened epiblastic tissue, omnipotent cells with strong mitotic activity, called the “primitive streak,” forms on the dorsocaudal surface of the embryonic disk. The primitive streak elongates in the cranial direction through cellular proliferation at its caudal end, causing the cranial end to thicken and become the primitive node (of Hensen). Meanwhile the primitive groove develops in the median plane; it terminates in a small depression called the primitive pit. A third germ layer, the embryonic mesoderm, begins to develop on approximately day 15. Starting from the primitive pit, mesoblastic cells are interposed between the ectoderm and endoderm along the primitive groove, initially forming a loose network and later fusing along the midline. The mesoblastic cells (embryonic mesenchyme) later differentiate into fibroblasts, chondroblasts, and osteoblasts. On day 16 additional cells migrate cephalad from the primitive node to form the head process, or notochordal process. On approximately day 18, portions of the notochordal process form the chordal plate, which infolds from its cranial end to form the actual notochord. At this time a small temporary channel forms between the yolk sac and amniotic cavity, the neuroenteric canal, which in turn is obliterated at the end of 4 weeks. The notochord is the central structure around which the spinal column develops and which induces the formation of the neural plate, which will give rise to the central nervous system. In later life, remnants of the notochord are present only in the nucleus pulposus of the intervertebral disks. The blastula and gastrula stages are followed by the development of the neural plate. The embryo at this stage is also called the “neurula.” At the end of 3 weeks of gestation, the notochord induces the transformation of superficial ectoderm to neural ectoderm, resulting in the formation of the neural plate or placode. The invagination process begins on day 17 as the lateral portions of the neural plate thicken and then fuse at the midline, resulting in closure of the neural tube (neurulation). This process occurs simultaneously in the cranial and caudal directions. There is still disagreement as to whether the fusion has one or multiple initiation points; this is important for understanding the embryology of certain malformations. The cranial end of the neural tube (anterior or rostral neuropore) closes on days 24 to 25, while the caudal end (posterior neuropore) closes on days 27 to 28. After closure of the neural tube, the neural and superficial ectoderm separate; the superficial ectoderm fuses in the midline and will form the dorsal skin ( ▶ Fig. 17.3). Fig. 17.3 Primary neurulation and disjunction disorders. Diagrammatic representation (from Jansen O, Stephani U. RRN Fehlbildungen und frühkindliche Schädigungen des ZNS. Stuttgart: Thieme; 2007). (a) In normal neurulation, the neural folds appose at the midline and fuse together. The superficial ectoderm separates from the neural ectoderm (disjunction, stage III) to create an intact skin covering. (b) In premature disjunction, contact between the mesenchyme and primitive ependyma leads to abnormal differentiation into fat and the development of spinal lipoma. (c) A focal failure of disjunction results in an epithelialized connection between the skin and neural tube. This mechanism is believed to be responsible for dermal sinus formation. Some ectodermal cells at the edge of the neural plate lose contact with neighboring cells during neurulation. When the neural tube separates from the superficial ectoderm, these cells migrate between the structures from both sides and form a flat layer of cells called the neural crest. This coherent layer soon separates again into two lateral cords and later forms the spinal ganglia and the sensory ganglia of the cranial nerves. Ingrowth of mesenchyme occurs between the skin and neural tube and will go on to form the meninges, paraspinal muscles, and posterior vertebral elements. Current theory holds that the posterior neuropore is most likely located at the S3 level. The more caudal portions of the spinal column and spinal cord are formed by day 48 of gestation, after the completion of primary neurulation. They arise from undifferentiated, omnipotent cells of the “caudal cell mass,” which in turn arises from fusion of the neural ectoderm and caudal notochord. On approximately day 30, cysts and vacuoles form within the caudal cell mass and fuse to form a tubular structure. This canal gains attachment to the primary neural tube. The process of retrogressive differentiation begins on day 38 and is characterized by regression of the caudal cell mass and caudal canal through programmed cell death (apoptosis) and further differentiation. It is very likely that this caudal segment gives origin to the distal conus medullaris, filum terminale, and terminal ventricle, which is obliterated in later life. Note Spinal malformations are a result of developmental abnormalities that occur between the third and sixth weeks of gestation. Spinal malformations are a very heterogeneous group of disorders that may occur in isolation but are very often found in a variety of combinations. We can understand them only by understanding their embryology, so it is logical to base their diagnostic classification on normal development ( ▶ Table 17.1). For everyday imaging practice, however, it is better to base the classification of spinal anomalies on clinical criteria. This enables us to make key preliminary differential diagnostic considerations leading to a more rational and efficient diagnostic protocol for affected children, who are usually sedated for examination ( ▶ Table 17.2). Disturbances of embryonic development Malformations Disorders of gastrulation Segmentation disorders of the spinal column Dorsal–enteric fistula Enterogenous cyst Diastematomyelia Caudal regression syndromes Segmental spinal dysgenesis Disorders of primary neurulation Spina bifida occulta Myelocele, myelomeningocele Lipomyelocele, lipomyelomeningocele Intradural lipoma Dermal sinus Dermoid Cervical myelocystocele Combined disorders of gastrulation and primary neurulation Hemimyelocele, hemimyelomeningocele Disorders of secondary neurulation and retrogressive differentiation Lipoma of the filum terminale Tight filum terminale Terminal myelocystocele Sacrococcygeal teratoma Disorders of unknown cause Meningocele Anterior sacral meningocele Clinical presentation Malformations Open spinal dysraphism Myelocele, myelomeningocele Hemimyelocele, hemimyelomeningocele Closed spinal dysraphism with subcutaneous swelling Lipomyelocele, lipomyelomeningocele Myelocystocele Meningocele Sacrococcygeal teratoma with cutaneous stigmata Dermal sinus Dorsal enteric fistula Diastematomyelia without a visible lesion Simple vertebral arch defect Segmentation anomalies of the spine Tight filum terminale Lipoma of the filum terminale Intradural lipoma Dermoid Enterogenous cyst Caudal regression syndromes Segmental spinal dysgenesis Pathology Open spinal dysraphisms are a group of anomalies that fall under the heading of primary neurulation disorders. They result from incomplete closure of the neural tube and associated incomplete fusion of the superficial ectoderm to form the integument. The failure of neural tube closure at this level leads to incomplete differentiation of the neural tissue and thus to persistence of the embryonic neural plate (placode). Additionally, mesenchymal cells cannot migrate between the neural plate and ectoderm, resulting in anomalous development of the posterior vertebral elements and back muscles. Thus, incomplete closure of the vertebral arch is not the cause of open spinal dysraphism but rather is the result of incomplete neural tube closure. Patients with a vertebral arch defect almost always have a Chiari II malformation, in which the intracranial changes are attributed to altered cerebrospinal fluid (CSF) dynamics arising from a failure of neural tube closure. Epidemiology The incidence of open spinal dysraphism is approximately 0.6:1000 live births. Lately the incidence has fallen significantly in developed countries as a result of conscientious folic acid replacement during pregnancy. MRI findings, treatment The clinical presentation is usually unmistakable, and surgical repair is performed during the first 48 hours of life due to the risk of infection. In highly selected cases where MRI is done postnatally because of equivocal clinical or ultrasound findings, it should be performed under sterile conditions in the prone or lateral decubitus position. There have been reports of prenatal surgical correction, but its benefits are controversial. The alleged advantages are less damage to the neural placode from mechanical and chemical factors and a positive effect on the intracranial changes of Chiari II malformation owing to the normalization of CSF dynamics. Prenatal MRI is often performed as an adjunct to ultrasound so that the extent of the malformations can be more accurately assessed. The most commonly diagnosed entity in this group is myelomeningocele ( ▶ Fig. 17.4), which occurs predominantly at the lumbosacral level but may occur anywhere along the spinal canal. Fig. 17.4 Myelomeningocele. Sagittal T1w image a few hours after birth demonstrates a lumbosacral myelomeningocele. The elongated spinal cord terminates in the neural placode, which forms the surface of the protruding sac. Clinical manifestations Physical examination reveals a reddish swelling in the midline that blends laterally with normal skin on both sides. MRI findings In most cases the sac contains the terminal cord or cauda equina structures. The structures in the sac may terminate in the neural placode, which also forms the outer surface. The neural placode is covered by a vascular plexus that normally forms the inner ependymal lining of the central canal. Tethering is always present with a lumbosacral myelomeningocele. Myelomeningoceles located at a higher level often show normal spinal cord caudal to the defect, which supports the theory that neurulation occurs simultaneously at different levels. Myeloceles are very rare and differ from myelomeningoceles only in the absence of an enlarged subarachnoid space. They may present clinically with an exposed neural placode and little or no swelling. Incomplete neural tube closure is often associated with a gastrulation disorder. A combination of myelo(meningo)cele with diastematomyelia is found in approximately 25% of all myelomeningocele patients but usually occur at different levels. Occurrence at the same level may lead to the very rare hemimyelomeningocele, which is generally limited to one side. Clinically, a swelling is noted adjacent to the midline and is accompanied by a unilateral neurologic deficit, which suggests the correct diagnosis. Almost all cases are associated with a Chiari II malformation. As a rule, the neurologic deficit from a myelomeningocele is stable following surgical repair. Neurologic deterioration (after the exclusion of decompensated hydrocephalus) is suspicious for a postoperative complication. The main postoperative causes of neurologic deterioration are: Retethering by scar tissue. Tethering by a second malformation not detected prior to surgery. Constricting dural ring. Ischemia due to vascular compression. Compression of the spinal cord by a dermoid, epidermoid, or arachnoid cyst. Development of hydromyelia. The most frequent complication, retethering by scar tissue ( ▶ Fig. 17.5), is also the most difficult to diagnose. Besides ultrasonography, which can be done in patients of all ages with a vertebral arch defect, ▶ MRI in the prone position has become an established routine technique for the direct detection of adhesions. Fig. 17.5 Complications of myelomeningocele. (a) Hydromyelia and tethering: The sagittal T2w image was taken a few weeks after removal of a sacral myelomeningocele. The spinal cord is still stretched and tethered at the level of the former sac. Hydromyelia involves almost the full length of the cord, with dilatation of the central canal most pronounced at the C4–T1 vertebral levels. Incidental finding: CSF pulsation artifacts at typical sites posterior to the thoracic cord. (b) Hydromyelia and an associated Chiari II malformation: status after surgical repair of a lumbar myelomeningocele. Sagittal T1w image of the neurocranium shows findings typical of a Chiari II malformation, with a shunt already in place. The cerebellum appears crowded in the small posterior fossa, with a low attachment of the tentorium. The tonsils have herniated into the spinal canal. Extensive hydromyelia of the cervical cord with fibrous septa is noted as an important “incidental finding” on cranial MRI. (c) Sharp-angled kyphosis of the lumbar spine following surgical treatment of lumbar myelomeningocele. The angulation resulted from instability due to a vertebral arch defect at that level, with associated blocking and wedging of the L3 and L4 vertebral bodies and thoracic-level paralysis. Sagittal image shows the stretched spinal cord tethered at the level of the former sac, with irregular cauda equina fibers. A constricting dural ring can be identified as a circumscribed, hourglass-shaped deformation of the spinal cord. Ischemic injury appears as a segmental narrowing of the cord with a somewhat greater craniocaudal extent. Dermoids and epidermoids may be congenital malformations or may develop after the surgical repair of dysraphic anomalies due to the inclusion of tissue rests or omnipotent cells of underdifferentiated tissue. Dermoids sometimes show a characteristic fat signal on MRI, although their signal characteristics, like those of epidermoids, may be highly variable. Tips and Tricks Both ▶ dermoids and epidermoids are often isointense to CSF in T1w and T2w sequences, making them difficult to discern. FLAIR and diffusion-weighted imaging (DWI) sequences should therefore be used to investigate an indeterminate mass. These sequences can demonstrate dermoids and epidermoids as solid tissue structures, which are almost always hyperintense to CSF on FLAIR images and show restricted diffusion on DWI in relation to CSF. Arachnoid cysts may also be congenital or may be acquired postoperatively in the form of arachnoid septations. As fluid-filled structures, they are isointense to CSF in all sequences and can often be diagnosed only indirectly by their mass effect. The development of hydromyelia is quite common, its incidence correlating closely with the control of hydrocephalus. It results from the reflux of CSF from the fourth ventricle into the central canal, caused by altered CSF dynamics after sac removal with concurrent narrowing of the foramen magnum due to downward displacement of the cerebellar tonsils. Hydromyelia is easily detected by MRI, in which the dilatation of the central canal is always located above the placode, usually at the cervicothoracic level and frequently segmented. Without treatment, hydromyelia may quickly incite the development of scoliosis or sharp-angled kyphosis. Clinical symptoms are segmental weakness and atrophy with loss of reflexes, sensory disturbances, and burning pain. These malformations present clinically with a soft, mobile swelling that is usually situated on the midline. It is typically found at the lumbosacral level and less commonly in the neck. The swelling is covered by skin, which may show local dystrophic changes. Epidemiology Lipomyeloceles ( ▶ Fig. 17.6) or the much rarer lipomyelomeningoceles ( ▶ Fig. 17.7) present as a lumbosacral swelling above the anal fissure that may extend asymmetrically into one buttock. They are decidedly common malformations that account for 76% of all spinal lipomas. Fig. 17.6 Lipomyelocele. Lipomyelocele with subcutaneous swelling at a typical lumbosacral site in a 3-year-old boy. The lipoma extends through the posterior vertebral arch defect from the neural placode, which is rotated slightly to the left, into the subcutaneous fat (from Jansen O, Stephani U. RRN Fehlbildungen und frühkindliche Schädigungen des ZNS. Stuttgart: Thieme; 2007). (a) Sagittal T2w image. (b) Sagittal T1w image. (c) Sagittal fat-saturated T1w image after contrast administration. (d) Axial T1w image. (e) Axial T1w image in a different plane. (f) Axial T1w image in a different plane. (g) Axial T2w image. Fig. 17.7 Lipomyelomeningocele in a 10-day-old girl with a soft lumbosacral swelling. The typical findings of a lipomyelocele are accompanied by cystic expansion of the dural sac into the subcutaneous fat. Subcutaneous fat extends continuously into the anterior spinal canal. The conus medullaris is fused with the intradural part of the lipoma. Fat-saturated images show tethering of the conus medullaris and filum terminale at the caudal end of the dural sac, which is not covered by bone. (a) Sagittal T2w image. (b) Sagittal T1w image. (c) Sagittal fat-saturated T1w image. (d) Axial fat-saturated T1w image. Clinical manifestations While these dysraphisms are detectable at birth, initial symptoms generally appear after 6 months in the form of muscle weakness, sensory disturbances, or neurogenic bladder dysfunction. Like other body fat, lipomas may “grow” during the first years of life or “shrink” in response to general weight loss. Pathology Lipomyelocele and lipomyelomeningocele are based on a neurulation disorder in which the cutaneous and neural ectoderm separate prematurely at a time when neural tube closure is not yet complete. Embryonic mesenchyme becomes interposed between the cutaneous and neural tissue, migrating through a still-patent gap in the dorsal neural tube into the central canal. Under the influence of the embryonic ependyma, the mesenchyme is transformed to fatty tissue that prevents complete neural tube closure and the normal differentiation of mesenchymal tissue to meninges, muscle, and posterior vertebral elements. The intradural lipoma extends through the meningeal and bony defect into the subcutaneous fat. If focal enlargement of the subarachnoid space is also present, the dysraphism is called a lipomyelomeningocele. MRI findings MRI will generally demonstrate an attachment between the lipoma and neural tissue (placode) in the lumbosacral region. The placode–lipoma interface is intraspinal with a lipomyelocele and is usually extraspinal with a lipomyelomeningocele. The structures of the conus medullaris and cauda equina are fused with the lipoma, with fatty tissue extending to the central canal. Nerve roots are often embedded asymmetrically within the fat, which may extend throughout the intra- and extradural space. A tethered cord is always present, and there is associated hydromyelia in 25% of cases. Bony, muscular, or vascular structures may be found within the lipoma as dysraphic hamartomas. Treatment In many cases, surgery can only reduce the extra- and intraspinal portions of the lipoma. Complete separation of the conus is rarely feasible due to the complex attachment between the neural tissue and lipoma. Clinical manifestations The very rare cervical myelocystocele is a soft midline swelling, covered by normal skin and usually located at the level of the cervicothoracic junction or several segments lower. In contrast to myelomeningocele, children with a myelocystocele are neurologically normal. Pathology There is disagreement concerning the pathogenesis of myelocystocele. Presumably it involves a neurulation disorder in which the initially large central canal fails to undergo sufficient segmental retraction and remains dilated. This leads to rupture of the adjacent dorsal mesenchyme, resulting in a vertebral arch defect that allows herniation of posterior cord tissue and the enlarged central canal. The liquid contents of the myelocystocele communicate with the central canal, but septations may also be present. Terminal myelocystoceles are extremely rare anomalies that present with a sacrococcygeal subcutaneous swelling, which is completely covered by skin. The underlying embryologic disorder involves a disturbance of secondary neurulation and retrogressive differentiation. This is presumed to alter CSF dynamics, leading to abnormal persistence and cystic dilatation of the terminal ventricle ( ▶ Fig. 17.8). This is another case in which destruction of the embryonic mesenchyme causes a vertebral arch defect with herniation of posterior cord tissue and an enlarged terminal ventricle. Fig. 17.8 Terminal myelocystocele in a mature newborn with subcutaneous lumbosacral swelling. A cyst of CSF signal intensity bulges into the subcutaneous tissue through a large spina bifida defect. The central canal shows increasing dilatation from above downward and, after the cord passes through the meningocele, ends in a large terminal cyst (from Jansen O, Stephani U. RRN Fehlbildungen und frühkindliche Schädigungen des ZNS. Stuttgart: Thieme; 2007). (a) Sagittal T2w image. (b) Sagittal T2w image in a different plane. (c) Sagittal T1w image. (d) Axial T2w image. (e) Axial T1w image.
17.2 Embryology
17.2.1 Gastrulation


17.2.2 Primary Neurulation

17.2.3 Secondary Neurulation and Retrogressive Differentiation
17.3 Classification
17.4 Open Spinal Dysraphisms
17.4.1 Myeloceles and Myelomeningoceles

17.4.2 Hemimyeloceles and Hemimyelomeningoceles
17.4.3 Postoperative Complications

17.5 Closed Spinal Dysraphisms
17.5.1 Closed Spinal Dysraphisms with Subcutaneous Swelling
17.5.1.1 Lipomyeloceles and Lipomyelomeningoceles


17.5.1.2 Myelocystoceles

Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


