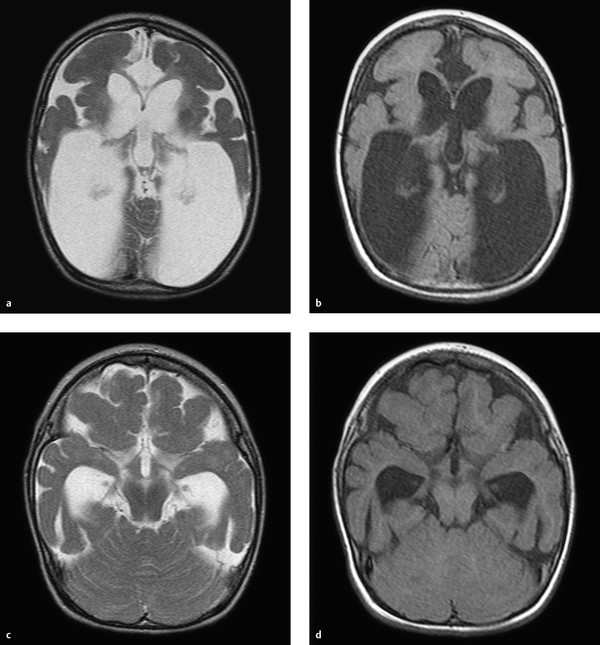
To understand malformations of the brain, the reader should be familiar with the critical processes that occur during embryonic development. This chapter focuses on the developmental processes that are essential for understanding important malformations and developmental abnormalities. In approximately the third week of gestation, a flat neural plate forms which will give rise to the entire central nervous system (CNS). The edges of the neural plate thicken and elevate to form the neural folds, which converge toward the midline and fuse to form the neural tube. The brain develops from the cranial part of the neural plate, and the spinal cord from the caudal part. Initially the neural tube is still open at its upper and lower ends. These openings are called the anterior and posterior neuropores. Following closure of the anterior neuropore, the three primary brain vesicles develop: the prosencephalon (forebrain), mesencephalon (midbrain), and rhombencephalon (hindbrain). The prosencephalon gives rise to the two cerebral vesicles and two optic vesicles. Between the cerebral vesicles is the lamina terminalis. Taken together, the cerebral vesicles and lamina terminalis make up the telencephalon or “endbrain.” Finally the lateral ventricles are formed from the cerebral vesicles. The basal ganglia develop at the base of the lateral ventricles, and the rudimentary cerebral cortex develops around their remaining walls. In approximately the seventh week of gestation, the dorsal part of the lamina terminalis condenses to form the rudiments of the corpus callosum and commissures. Its ventral part gives rise to the primitive meninx, which is the precursor of the meninges. Development of the corpus callosum follows a very definite sequence: The first structure to form is the posterior part of the genu, followed by the body and anterior part of the genu and finally the splenium. The rostrum of the corpus callosum forms last. We can simplify this scheme by stating that, except for the rostrum, the corpus callosum develops in an anterior-to-posterior direction. This principle is important for understanding hypogenesis of the corpus callosum. The lamina terminalis, which increasingly stretches and thins during callosal development, finally gives rise to the septum pellucidum. Roughly synchronous with the development of the corpus callosum, the neurons that will give rise to the cerebral cortex form in the immediate subependymal layer. This zone, which borders directly on the lateral ventricles, is called the germinal matrix zone. It is an area of high metabolic activity during fetal brain development. Note During fetal development, neurons form in the germinal matrix zone and migrate from there to the cortex. The matrix zone is highly susceptible to injurious effects during fetal development, such as cellular hypoxia due to deficient blood flow. The neurons migrate from the germinal matrix zone to the surface of the brain. This migration begins in approximately the eighth week of gestation and proceeds radially in a centrifugal pattern. Once the neurons have reached the brain surface, the process of cortical organization begins—the cortical cells are arranged in layers. When the brain begins to develop, its surface is completely smooth. Infolding of the sylvian fissure begins in approximately the fourth month, at which time the brain is shaped like a figure 8. The cortex is still quite thin initially, as only some of the neurons have reached the surface. Additional gyri and sulci start to develop in approximately the 20th week, appearing first in the parieto-occipital region and cingulum. The gyri and sulci are not completely formed until the pregnancy reaches term, although the sulci of newborns are not as deep as in older children or adults. In evaluating developmental abnormalities of the cortex, it should also be noted that development of the cerebral gyri proceeds in stages. The brain is not completely smooth during embryonic and early fetal development, but shows a “physiologic lissencephaly.” Van der Knaap (1996) divided the gyration process into five stages according to the following timetable: Stage 1: Before week 32. Stage 2: Weeks 33–34. Stage 3: Weeks 35–37. Stage 4: Weeks 38–41. Stage 5: After 41 weeks. Abnormalities of gyration may occur in any of these five stages. The white matter of the brain is initially unmyelinated in the fetus. Myelination begins in approximately the fifth gestational month and proceeds relatively slowly until term. In an infant delivered at term, generally only the posterior limb of the internal capsule, portions of the medulla oblongata, the posterior midbrain, and portions of the cerebellar peduncles are myelinated. After birth, myelination should proceed continuously in the first 2 years of life according to a fixed timetable. Note The only myelinated brain structures at term are the posterior limb of the internal capsule, portions of the medulla oblongata, the posterior midbrain, and portions of the cerebellar peduncles. The pituitary develops differently from other brain structures. The infundibulum is formed from part of the diencephalon and gives rise to the pituitary stalk and posterior lobe, but the anterior lobe of the pituitary develops from the Rathke cleft, an ectodermal outpouching of the stomodeum rostral to the pharyngeal membrane. This arrangement helps us understand the pathophysiology of a pharyngosellar pituitary and Rathke cleft cysts. The metencephalon arises from the anterior part of the rhombencephalon. As embryonic development proceeds, the anterior part of the metencephalon gives rise to the pons and middle cerebellar peduncle, while the posterior part gives rise to the cerebellum. The posterior lips of the rhombencephalon form at the start of cerebellar development and converge to form the cerebellar plate. After 3 months gestation the vermis and cerebellar hemispheres can be identified as separate structures. The nodulus and flocculus are differentiated shortly thereafter. The medullary velum develops from the cover plate of the fourth ventricle. To correctly evaluate and classify abnormalities of cortical development, it is helpful to know the principles governing the development of the cortical ribbon. As described above, the neurons are initially formed in the germinal matrix zone, then migrate radially to the brain surface (neuronal migration), where they form a layered arrangement (cortical organization). Barkovich et al (1996) devised a classification based on this concept which divided cortical malformations into three categories: Disorders of neuronal proliferation: i.e., the development of neuronal stem cells in the germinal matrix zone. Disorders of neuronal migration: i.e., migration of the neurons from the matrix zone to the brain surface. Disorders of cortical organization: i.e., the arrangement of the neurons in the cortical ribbon. This classification was revised at regular intervals and modified in accordance with new discoveries, often based on molecular genetic studies. In 2012, Barkovich et al introduced the following classification system for cortical malformations: Group I: Malformations due to abnormal neuronal and/or glial proliferation or apoptosis (apoptosis = programmed cell death): Group I.A: Disorders due to reduced proliferation or accelerated apoptosis—congenital microcephalies. Group I.B: disorders Due to increased proliferation or decreased apoptosis—megalencephalies. Group I.C: disorders due To abnormal proliferation—focal and diffuse dysgenesis and dysplasia. Group II: malformations due to abnormal neuronal migration: Group II.A: abnormal neuroependymal initiation of migration (i.e., abnormal initial migration in the subependymal germinal matrix zone)—predominantly periventricular (subependymal) heterotopias. Group II.B: generalized abnormalities of transmantle migration (i.e., migration from the subependymal germinal matrix zone to the cortex)—mainly lissencephalies. Group II.C: localized abnormalities of transmantle migration—mainly subcortical heterotopias. Group II.D: abnormalities of terminal migration, i.e., involving the “last step” in the migratory pathway—mainly cobblestone lissencephalies, including less severe forms in fetal alcohol syndrome. Group III: malformations due to abnormal postmigrational development: Group III.A: polymicrogyria and schizencephaly (caution: polymicrogyria occurs in Groups III.A, III.B, III.C, and III.D). Group III.B: polymicrogyria without clefts or calcifications. Group III.C: focal cortical dysplasias. Group III.D: postmigrational microcephaly. This chapter deals mainly with developmental abnormalities of the cortex that are relevant to everyday practice or are particularly helpful for illustrating the principle of cortical development. Note The etiology of cortical malformations is often uncertain, but it is important to consider genetic causes, intrauterine ischemia or infection, and intrauterine toxic exposure. Microcephaly with a simplified gyral pattern (MSG) is a group I malformation, meaning that it is secondary to abnormal neuronal proliferation. MSG, also called “microlissencephaly,” is characterized by the reduced proliferation of neuronal stem cells or increased apoptosis, or an increase in programmed cell death (apoptosis). By definition, microcephaly is present when the head circumference is at least 2 standard deviations below mean. Extreme microcephaly is present if the head circumference is more than 3 standard deviations below mean. Note If the head circumference at birth is less than 2 standard deviations below mean and severe microcephaly develops during the first 2 years of life, the disorder is classified in Group III. Patients with MSG not only show a decrease in neurocranial volume but also have an abnormally simplified gyral pattern. The number of gyri and sulci are significantly reduced. Typically only a few shallow sulci are present ( ▶ Fig. 9.1). Fig. 9.1 MSG. Microcephaly with a simplified gyral pattern in an 18-month-old girl. There is associated ex-vacuo expansion of the white matter. (a) Axial T2w image. (b) Axial T1w image. (c) Axial T2w image in a different plane. (d) Axial T1w image in a different plane. The MSG group is currently subdivided into seven subgroups: MSG group 1: This type usually follows an uneventful pregnancy. At birth, however, the infant is found to have a reduced head circumference and soon thereafter will generally exhibit a developmental delay. MRI shows a decreased cerebral volume with reduced gyri and sulci. The cortical ribbon itself appears normal. MSG group 2: Patients with this malformation usually show a spastic increase in muscle tone immediately after birth, which is often a breech delivery. Early onset of epileptic seizures is common. The MRI appearance is the same as in group 1 , but myelination is often delayed in infants and small children. MSG group 3: This malformation usually follows an uneventful pregnancy, but newborns quickly exhibit seizures and diminished reflexes. MRI shows fewer gyri and sulci compared with groups 1 and 2. It is also common to find periventricular gray-matter heterotopias and arachnoid cysts. MSG group 4: This type is associated with additional significant pre- or postnatal disorders such as arthrogryposis multiplex congenita, polyhydramnios, or atresia of the jejunum. The MRI findings are similar to those in group 1; myelination is appropriate for age. MSG group 5: This type is characterized by extreme microlissencephaly with antenatal hypotonia and epileptic seizures. The brain shows a simplified gyral pattern with a maximum of five gyri in each hemisphere. MSG group 6: This anomaly is characterized by complete or nearly complete agyria (absence of gyration). Many patients also have agenesis of the corpus callosum, cerebellar hypoplasia, or subependymal heterotopias. MSG group 7: This anomaly is also called the “leukodysplasia, microcephaly, and cerebral malformation syndrome.” This recently described syndrome has been linked to a mutation on chromosome 2p16. Affected children have severe microcephaly, develop refractory epilepsy, and generally do not survive past 3 years of age. There are various syndromes in which both polymicrogyria and megalencephaly are present. These syndromes include: M-CMTC: Macrocephaly, cutis marmorata, and teleangiectatica congenita. MPPH: Macrocephaly, polymicrogyria, polydactyly, and hydrocephalus. MCAP: Macrocephaly, capillary malformation, and polymicrogyria. These syndromes are associated with varying degrees of polymicrogyria. The abnormal gyri often show a perisylvian distribution, but this is not always the case. Typically there is progressive ectopia of the cerebellar tonsils due to megalencephaly, resulting in a pseudo-Chiari malformation ( ▶ Fig. 9.2). Fig. 9.2 M-CMTC syndrome. (a) Sagittal T2w image shows a typical pseudo-Chiari malformation with caudal displacement of the cerebellar tonsils due to the increased brain volume. (b) Axial T2w image demonstrates focal polymicrogyria with patchy white-matter hyperintensities. Hemimegalencephaly is no longer listed in group I.B because dysmorphic cells have been found in patients with the disorder. For simplicity, however, it will be discussed in this section. Pathology Hemimegalencephaly is a relatively complex disorder in which all or part of one hemisphere undergoes hamartomatous changes, generally accompanied by enlargement of the affected area. The primary disorder is believed to be an abnormal proliferation of neuronal stem cells, but abnormalities of migration and cortical organization also occur. Clinical manifestations Most cases take a very severe course. Patients generally present in early childhood with epileptic seizures that show little or no response to medical therapy. MRI findings MRI usually shows enlargement of the affected hemisphere. The ipsilateral lateral ventricle may also be enlarged, which differs from the findings in neoplastic and other diseases. This is not always the case, however, and there are case reports in which the lateral ventricle of the affected hemisphere was not enlarged. The affected hemisphere may show heterotopias due to migration disorders and pachygyria or polymicrogyria due to abnormal cortical organization. Gliosis is also a relatively common finding. Patchy, inhomogeneous white-matter hyperintensity is often present. The cortical ribbon is thickened and dysplastic. The gray–white matter junction is often indistinct ( ▶ Fig. 9.3). Fig. 9.3 Hemimegalencephaly. Axial T2w images show markedly decreased volume of the right hemisphere with enlargement of the ipsilateral lateral ventricle. The images also show extensive heterotopia and abnormal gyration (with kind permission of Dr. A. Seitz, Department of Pediatric Neuroradiology, Heidelberg University Hospital). (a) Axial T2w image. (b) Axial T2w image (next slice after a). (c) Axial T2w image (next slice after b). Clinical manifestations and pathology Focal cortical dysplasias (FCDs) are a frequent cause of epileptic seizures in children. They may also cause epilepsy that is not manifested until adulthood. FCDs are divided into several subtypes. It is likely that FCDs of type II represent disorders of neuronal proliferation. FCD type IIa contains dysmorphic neurons, while FCD type IIb contains balloon cells (a term used in histopathology for cells that are large and have abundant cytoplasm; they are believed to be neuronal stem cells that have not differentiated). Tubers in the setting of tuberous sclerosis resemble FCD type IIb in their histopathologic features. Focal transmantle dysplasia, called also transhemispheric FCD, has the same histopathology as FCD type IIb. MRI in this disorder shows a streak of dysplastic cells running from the subependymal germinal matrix zone through the entire hemisphere to the brain surface. Most affected patients present with epileptic seizures, which may have a focal onset. MRI findings FLAIR and T2w sequences in FCD type II usually show high signal intensity in the subcortical white matter ( ▶ Fig. 9.4). Not infrequently, the affected gyri have a somewhat “swollen” appearance. In infants less than 1 year old, the lesion is often hyperintense in T1w images and hypointense in T2w images relative to the surrounding, still-unmyelinated white matter. Only later do we find the typical subcortical hyperintensity in T2w and FLAIR sequences. After age 1 to 2 years, FCDs of type II are sometimes difficult to discern as a result of myelination processes. On the whole, type II FCDs have an MRI appearance similar to the tubers in tuberous sclerosis, and sometimes show a dysplastic streak running from the former subependymal matrix zone to the cortical surface. The streak usually has a linear or funnel shape. The adjacent cortex is also dysplastic as a rule. The gray–white matter junction is indistinct. Because the dysplastic zone may be quite narrow, it is not always easy to identify on MRI. High-resolution sequences are recommended, as they can provide good gray–white matter differentiation with optimum spatial resolution ( ▶ Fig. 9.5). The transhemispheric streak appears to be specific for FCD type II but is not always present. Fig. 9.4 FCD type II. Axial FLAIR image shows focal cortical–subcortical hyperintensity in the right hemisphere. Fig. 9.5 Focal transmantle dysplasia. A broad streak of dysplasia runs from the ventricular ependyma to the cortical surface in a 19-year-old woman. Next to it is a cortical ribbon abnormality with dysplastic components and polymicrogyria. (a) Axial FLAIR image. (b) Axial T1w image. Heterotopias are the “classic” disorder of neuronal migration. An interruption of normal neuronal migration from the subependymal germinal matrix zone to the cortex results in gray matter at abnormal locations. The following two forms are distinguished: Periventricular (subependymal) nodular heterotopias: These are disorders of Group II.A caused by abnormal neuroependymal initiation of migration (i.e., abnormal initial migration of neurons from the subependymal zone). Focal subcortical heterotopias: These are disorders of Group II.C, i.e., localized abnormalities of transmantle migration. The neurons started their migration to the cortical surface but did not arrive. The clinical significance of heterotopias is highly variable. If a heterotopia is present with no additional brain malformations, most patients will develop normally. Heterotopias may present clinically with epileptic seizures, but they may also be noted as purely incidental findings. If periventricular heterotopias almost completely line the ventricle wall, an X-linked mode of inheritance should be considered. Autosomal recessive inheritance has also been described. Periventricular heterotopias are in direct contact with the ventricular ependyma. Most have a rounded shape, but occasionally they are oval-shaped, usually with their long axis parallel to the ventricle. Subependymal heterotopias mainly require differentiation from subependymal nodules in tuberous sclerosis. Heterotopias are isointense to the cortical ribbon in all MRI sequences ( ▶ Fig. 9.6), whereas subependymal nodules in tuberous sclerosis are not isointense to the cortex. Also, they often contain calcifications which are best demonstrated by CT. Fig. 9.6 Subependymal heterotopias. Heterotopic subependymal gray matter in a 13-year-old boy. The heterotopias are isointense to the cortical ribbon in all sequences. (a) Axial T2w image. (b) Axial T2w image in a different plane. (c) Axial FLAIR image. (d) Coronal T1w inversion-recovery image. These heterotopias, unlike the subependymal heterotopias, consist of neurons that began their migration to the cortical surface but were retained in the white matter. As a result, cell clusters are found within the white matter that are isointense to cortex in all sequences. They may be isolated but are usually multiple. They have a round to elongated shape. Elongated heterotopias usually show a radial arrangement matching the radial, centrifugal migratory pathway of the neurons. The associated cortical ribbon may show dysplastic changes or may be thinned. Heterotopias can be distinguished from other white-matter lesions such as intra-axial tumors by noting their isointensity to cortex in all sequences. Moreover, heterotopias never show perifocal edema. Another helpful differentiating criterion is possible dysplasia of the associated cortex ( ▶ Fig. 9.7). Fig. 9.7 Focal subcortical heterotopias. Heterotopic gray matter (arrows in a,d) is visible within the gray matter in a 14-year-old boy. The heterotopias are isointense to cortex in all sequences. (a) Axial T2w image. (b) Axial T1w image. (c) Axial FLAIR image. (d) Sagittal T2w image. Lissencephalies are Group II.B disorders, i.e., abnormalities of transmantle migration. The hallmark of lissencephaly (“smooth brain”) is an absence or significant reduction of physiologic gyration. Another term for the lissencephaly spectrum is “agyria–pachygyria complex.” Lissencephaly may be complete (agyria) or incomplete (pachygyria). Localized pachygyria may also occur. Pathology The cause of classic lissencephalies may be a chromosomal abnormality, especially of the X chromosome and chromosome 17. An abnormality of chromosome 17 combined with characteristic facies is known as Miller–Dieker syndrome. When X-linked lissencephaly is present in a boy, the mother often has a laminar heterotopia, underscoring the close relationship of these two processes. In recent years mutations in the TUBA1A gene have also been described that are responsible for classic lissencephalies and especially for lissencephalies with cerebellar hypoplasia. Clinical manifestations Children with disorders of the lissencephaly spectrum usually present clinically with seizures, very often accompanied by developmental delay. The severity of symptoms depends on the extent of the disorder. MRI findings Complete lissencephaly: MRI in complete (classic) lissencephaly shows a smooth brain that is devoid of gyri and sulci. The sylvian fissure is present in most cases, however, causing the brain to have a figure-8 appearance when imaged in axial sections (“figure-8 lissencephaly”). The inner cerebrospinal fluid (CSF) spaces are usually enlarged in patients with classic lissencephaly. Incomplete lissencephaly: This form is more common than complete, classic lissencephaly. Infolding of the brain surface has begun, but gyration remains incomplete. Zones of agyria may coexist with zones of pachygyria featuring broad gyri and shallow sulci. The cortical ribbon is usually thickened and has a smooth junction with the gray matter. This is an important differentiating feature from polymicrogyria. Pachygyria may also be one part of a spectrum, however, that includes agyria–pachygyria complex and polymicrogyria. As a result, it is not unusual to find areas of pachygyria coexisting with areas of polymicrogyria in the same patient ( ▶ Fig. 9.8). Fig. 9.8 Pachygyria and polymicrogyria. Pachygyria, with a thick cortical ribbon and simplified gyration, coexists with areas of polymicrogyria with numerous small gyri. (a) Axial T2w image (plane 1). (b) Axial T1w image (plane 1). (c) Axial FLAIR image (plane 1). (d) Axial T2w image(plane 2). (e) Axial T1w image (plane 2). (f) Axial FLAIR image (plane 2). Pathology Laminar heterotopias are also considered part of the lissencephaly spectrum. They are very often based on an underlying X-linked disorder and are more common in females than males. Sons of women with a laminar heterotopia will often have classic X-linked lissencephaly. Laminar heterotopias are caused by a diffuse arrest of neuronal migration from the subependymal germinal matrix zone to the brain surface. Some neurons do reach the hemisphere surface, however, forming a bandlike layer that creates a “double cortex” appearance at imaging. This may affect just one part of the cortical ribbon or the entire cortex. MRI findings MRI shows a laminar band of variable width that is located within the white matter and generally runs parallel to the ventricular surface. Like all heterotopias, the band is isointense to gray matter in all sequences. A zone of normal-appearing white matter may be interposed between the heterotopic band and the cortex or ependyma ( ▶ Fig. 9.9). Fig. 9.9 Laminar heterotopia. Bilateral bands of altered signal intensity in the white matter are isointense to cortex in all sequences. This case also shows focal subcortical heterotopias and cystic dilatation of the lateral ventricles. (a) Axial T1w image (plane 1). (b) Axial T2w image (plane 1). (c) Axial T1w image (plane 2). (d) Axial T1w image (plane 3). Cobblestone malformations are Group II.D disorders, i.e., abnormalities of terminal migration involving the “last step” in the migratory pathway, leading to a defective pial basement membrane. Cobblestone malformations are often associated with muscular dystrophies. The group of cobblestone lissencephalies is considered to include the relatively rare syndromes of the Walker–Warburg complex, Fukuyama muscular dystrophy, and muscle–eye–brain disease, congenital muscular dystrophies type 1C and 1D, and limb-girdle muscular dystrophies: Walker–Warburg syndrome: In this syndrome, a cobblestone malformation of the cortex is almost always accompanied by hydrocephalus and ocular malformations, usually microphthalmia, and often by malformation of the corpus callosum and cerebellum. Fukuyama muscular dystrophy: This disorder, seen almost exclusively in Japan and very rarely in Europe, leads to predominantly occipitotemporal cobblestone lissencephaly, polymicrogyria (usually frontal), and subcortical cysts. Myelination does not follow the normal timetable but proceeds in reverse order. Muscle–eye–brain disease: This disease, which occurs mainly in Finland, resembles Walker–Warburg syndrome because it includes ocular malformations. Polymicrogyria is characterized by an excessive number of abnormally small gyri. It is the opposite of pachygyria, in which the gyri are broad and few in number. Nevertheless, it is sometimes difficult to distinguish between these two entities because the many small gyri in polymicrogyria may appear as one large gyrus when imaged at low resolution or by CT. Polymicrogyria is listed in Groups III.A, III.B, III.C and III.D in the 2012 classification of Barkovich et al. On the whole, polymicrogyria is remarkably heterogeneous at both the microscopic and macroscopic levels. Pathology Group III.A disorders: These polymicrogyrias are associated with schizencephalic clefts and/or calcifications. They presumably result from abnormalities of intrauterine development caused, for example, by an intrauterine vascular insult or infection, especially a congenital cytomegalovirus (CMV) infection. Not infrequently, however, a cause cannot be identified. Group III.B disorders: These disorders are not associated with a cleft anomaly or calcifications. Their etiology may include genetic causes. Clinical manifestations Patients with polymicrogyria often present clinically with epileptic seizures. Development is often delayed in affected children. MRI findings An MRI diagnosis of polymicrogyria requires high-resolution sequences in three planes or volume sequences. Affected brain areas exhibit numerous small or very small gyri, which are generally isointense to other gray matter. The adjacent white matter occasionally shows increased T2w signal intensity. Affected areas may contain calcifications, especially if the cortical malformation was caused by an intrauterine infection (e.g., a CMV infection, ▶ Fig. 9.10). In contrast to pachygyria, the gray–white matter junction is wavy and not smooth. This is a helpful criterion for differentiating the two entities. With non-high-resolution sequences, it is often difficult to distinguish a thick cortex from numerous small gyri. Fig. 9.10 Polymicrogyria secondary to congenital CMV infection. Polymicrogyric changes are noted at various sites in the brain. The gray–white matter junction appears wavy in those areas. There are coexisting areas of pachygyria and white-matter gliosis. (a) Axial T2w image (plane 1). (b) Axial T1w image (plane 1). (c) Axial FLAIR image (plane 1). (d) Axial T2w image (plane 2). (e) Axial T1w image (plane 2). (f) Axial FLAIR image (plane 2). Tips and Tricks A developmental venous anomaly (DVA) is occasionally found adjacent to a zone of polymicrogyria. It simply reflects the developmental malformation of the cortex and does not require further investigation. Schizencephaly literally means “split brain.” The cleft extends completely through the hemisphere from the ependyma of the lateral ventricle to the brain surface. The cleft is always completely lined by cortical tissue, which is generally dysplastic. The brain areas bordering the cleft usually show abnormal gyration, often in the form of polymicrogyria. Schizencephaly may be of two types: open-lip or closed-lip. Open-lip schizencephaly: In this type the cleft walls are separate and create an open communication between the ventricles and outer CSF spaces. Closed-lip schizencephaly: In this type, gray matter-lined cleft walls are apposed to each other. Clinical manifestations Most affected patients present in early childhood with a developmental delay. Many children also experience epileptic seizures caused by the gray matter dysplasia. Most patients with open-lip schizencephaly also have motor deficits. With bilateral schizencephaly, the signs and symptoms are usually more severe; many of these patients are blind due to bilateral disruption of the visual pathway. MRI findings MRI in schizencephaly demonstrates a unilateral or bilateral cleft extending completely through the hemisphere. It is often located in the pericentral region, but this is not necessarily the case. Open-lip schizencephaly This anomaly is usually easy to identify. The cleft is completely filled with CSF. Open-lip schizencephaly can be distinguished from a tissue defect following an middle cerebral artery (MCA) infarction, for example, by noting the presence of a gray-matter lining, which is absent in a simple defect. With a broad open-lip schizencephaly, there will often be focal thinning and bulging of the adjacent calvarium, most likely due to chronically increased CSF pulsations in that region ( ▶ Fig. 9.11 and ▶ Fig. 9.12). Fig. 9.11 Unilateral open-lip schizencephaly. A gaping cleft extends completely through the left hemisphere, forming a connection between the inner and outer CSF spaces. The full length of the cleft is lined with gray matter. (a) Axial T2w image. (b) Axial T1w image. (c) Coronal T1w inversion-recovery image. Fig. 9.12 Bilateral schizencephaly. On the right side is a broad open-lip cleft with associated thinning and focal bossing of the calvarium. Closed-lip schizencephaly is present on the left side. (a) Axial T2w image. (b) Axial T1w image. (c) Axial FLAIR image. (d) Coronal T1w inversion-recovery image. Closed-lip schizencephaly Closed-lip schizencephaly may be more difficult to diagnose. The cleft walls are in apposition and the cleft is not filled with CSF. As a general rule, however, the ventricular surface will show a focal, nipple-shaped diverticulum at the base of the cleft. Because the cleft is always lined by gray matter in schizencephaly, an area of gray matter within the white matter will be seen bordering the diverticulum in closed-lip schizencephaly. The cleft in that area is usually easy to identify in higher-resolution T1w sequences. Unlike focal transmantle dysplasia, closed-lip schizencephaly has two apposed “lips” of dysplastic gray matter that can be identified as separate structures on close scrutiny. Occasionally we recommend the acquisition of high-resolution sequences in at least two planes to avoid missing a closed-lip schizencephaly and ensure that the full extent of the cleft and adjacent abnormal cortical organization can be accurately evaluated. The superficial part of the cleft is almost always bordered by abnormal gyration in the form of polymicrogyria. Pitfall Because schizencephaly is often bilateral, the contralateral hemisphere should always be closely examined. It is not uncommon for open-lip schizencephaly to coexist with closed-lip schizencephaly on the opposite side, which may be missed on cursory inspection. Schizencephaly also has a high association with other disorders of cortical organization, which may be present in one or both hemispheres. Therefore do not diagnose an isolated, unilateral schizencephaly before carefully examining all other portions of the neurocranium for possible associated anomalies (see Fig. 9.12). Tips and Tricks If schizencephaly is suspected, a T1w inversion-recovery sequence is helpful for detecting gray matter on the cleft walls. FCD type I and type III are classified as Group III.C disorders, i.e., malformations due to abnormal postmigrational development and thus involving stages of cortical development that follow neuronal migration. In contrast to ▶ FCD type II, the histopathology of FCD types I and III does not include dysmorphic neuronal cells or balloon cells. FCDs of type I are focal structural anomalies of the cortex and usually of the adjacent white matter. They may have a more radial (type Ia) or horizontal (type Ib) orientation; mixed types also occur (type Ic). Clinical manifestations and treatment Affected patients generally present clinically with epileptic seizures, usually of a focal type. Because surgery is a treatment option for focal seizures, it is important to make this diagnosis as accurately as possible. MRI findings Overall, FCDs of histologic type I have a very diverse MRI presentation. According to the literature, at least 50% of patients with FCD type I have normal MRI findings. Even in patients with MRI abnormalities, it is often difficult to detect FCDs. Images often show only circumscribed blurring of the gray–white matter junction ( ▶ Fig. 9.13). Especially if an epileptic surgical procedure is proposed, very high-resolution images should be obtained in multiple planes along with volume sequences. Given the very long acquisition times, it may be necessary to scan the patient under general anesthesia to ensure optimum image quality. Fig. 9.13 FCD type I. Focal expansion of the paramedian cortical ribbon is apparent on the right side. The dysplasia appears slightly hyperintense in the FLAIR image. (a) Axial T2w image. (b) Axial T1w image. (c) Axial FLAIR image. FCDs of type III are associated with other disorders and are subdivided on that basis: FCD type IIIa: Associated with hippocampal sclerosis. FCD type IIIb: Associated with epileptogenic tumors such as dysembryoplastic neuroectodermal tumor, gangliogliomas, or gangliocytomas. FCD type IIIc: Associated with vascular malformations. FCD type IIId: Associated with scarring due to porencephaly, trauma, inflammation, etc. Patients with FCD type III are usually older than patients with FCD type II at the time of diagnosis, and many are diagnosed in adulthood. Also, type III FCDs are more commonly located in the temporal lobe than other FCD subtypes. The cerebral commissures that cross the midline include the anterior commissure, the hippocampal commissure, and the corpus callosum itself. The commissures form during a critical period of embryonic development in which many other brain structures are formed. In the seventh gestational week, the lamina terminalis thickens to form the lamina reuniens. The hippocampal commissure forms in the 11th week, and development of the anterior corpus callosum begins in the 13th week. Note When evaluating malformations of the corpus callosum, always examine the commissures as well. In classic agenesis of the corpus callosum, the anterior commissure is generally small but present whereas the hippocampal commissure is usually absent. During the development of the corpus callosum, the first structures to form are the posterior portions of the genu, followed by the body and anterior part of the genu, and then the splenium. The rostrum is formed last. In simplified terms, we may say that the corpus callosum forms “from front to back” with the anterior parts preceding the posterior parts. This is important because in typical hypogenesis of the corpus callosum, the anterior parts of the corpus callosum are present while the posterior portions are absent ( ▶ Fig. 9.14). But if portions of the body or genu are absent while the splenium is intact, this is probably a result of secondary damage rather than a developmental anomaly. Rare exceptions to this rule are ▶ holoprosencephaly and the middle interhemispheric variant of holoprosencephaly (syntelencephaly). Fig. 9.14 Hypogenesis of the corpus callosum. Portions of the corpus callosum are absent due to a developmental anomaly (hypogenesis). (a) Sagittal T2w image. (b) Sagittal T1w image. In contrast to developmental anomalies of the corpus callosum due to hypogenesis or agenesis, secondary damage refers to a lesion or thinning of the corpus callosum that occurs after the corpus callosum has fully formed. A relatively frequent cause of secondary thinning in the peri-isthmic region is a posthypoxic insult to the immature brain, like that occurring in a preterm delivery. Damage to the crossing fiber tracts leads to peri-isthmic thinning of the corpus callosum; this can be so pronounced that it may appear as a complete defect in that region. Secondary insults to the corpus callosum may also result from leukodystrophy, infarction, or autoimmune disorders such as encephalomyelitis disseminata ( ▶ Fig. 9.15). Fig. 9.15 Secondary thinning of the corpus callosum. The sagittal T2w image (a) shows a markedly reduced volume of the anterior corpus callosum. The posterior portions appear normal. The thinning occurred secondary to an MCA and anterior cerebral artery (ACA) infarction on the right side (b) and is not a primary developmental anomaly. (a) Sagittal T2w image. (b) Axial T2w image. Note It is important to differentiate between malformations of the corpus callosum and secondary damage, because malformations have a completely different etiology. Accurate classification of the disorder is important in directing a targeted search for the cause and in parental counseling, which may include information on recurrence risk in future pregnancies. Clinical manifestations Agenesis or hypogenesis of the corpus callosum may be almost asymptomatic. Hypogenesis or even agenesis of the corpus callosum is occasionally detected incidentally during the MRI of an adult patient imaged for a different indication. In these cases the callosal malformation itself can be diagnosed clinically only by detailed psychological testing. But because the corpus callosum is formed during an important and highly active phase of brain development, additional brain malformations are very often present, in which case the patient will often be markedly symptomatic in early childhood. Depending on the severity of the disorder, children will often exhibit developmental delays as well as epileptic seizures in cases with associated cortical malformations. MRI findings Tips and Tricks Given the frequent association of corpus callosum anomalies with other brain malformations, the corpus callosum has an important role in diagnosing developmental abnormalities of the brain. Thus, particular attention should be given to the corpus callosum in every MRI examination of the neurocranium involving a possible brain malformation. Especially in children with developmental delay, always acquire at least one sagittal sequence to facilitate evaluation of the corpus callosum. There are various direct and indirect signs of complete or partial absence of the corpus callosum on MRI. As a rule, sagittal slices will clearly demonstrate any agenesis or hypogenesis of the corpus callosum in cases where the white matter is already myelinated. But characteristic signs of callosal dysgenesis can also be seen in axial and coronal images: Colpocephaly: A typical sign of callosal agenesis or hypogenesis in axial images is enlargement of the lateral ventricles predominantly affecting the occipital horns. The corpus callosum contains a dense arrangement of fibers that contribute greatly to the shape and stability of the lateral ventricles. If the corpus callosum is absent or deficient, the lateral ventricles will expand. This enlargement predominantly affects the occipital horns, where it is often compounded by associated aplasia or hypoplasia of the cingulum. The enlargement is less pronounced at the level of the frontal horns, because the caudate nucleus and lentiform nucleus also contribute to ventricular shape in that region. Disproportionate enlargement of the occipital horns of the lateral ventricles is called “colpocephaly.” Crescent-shaped frontal horns of the lateral ventricles: This configuration in the coronal plane results from medial indentation of the lateral ventricles by “Probst bundles.” These bundles form when axons that would otherwise cross the midline to form the corpus callosum are diverted, forming longitudinal tracts. Steerhorn-shaped bodies of the lateral ventricles: With corpus callosum agenesis, the bodies of the lateral ventricles are nonconvergent and form parallel “steerhorns” when viewed in axial or coronal section ( ▶ Fig. 9.16). Fig. 9.16 Complete agenesis of the corpus callosum. The lateral ventricles show dilatation of the occipital horns and parallelism of the ventricular bodies. Image (c) shows absence of the corpus callosum and cingulum. (a) Axial T2w image. (b) Axial T1w image. (c) Sagittal T2w image. In a neonate or infant whose white matter is still mostly unmyelinated, it may be difficult to diagnose absence of the corpus callosum, even in sagittal images. A helpful sign in these cases is when the sulci of the medial hemisphere open into the third ventricle in a radiating pattern, which is often the case due to eversion or absence of the cingulate gyrus. The gyri and sulci of the medial brain surface then run directly into the third ventricle, bypassing the cingulum and creating a radial pattern when viewed in sagittal section. ▶ Fig. 9.16c demonstrates this radiating sulcal pattern in an 8-month-old boy whose myelination is still incomplete. Agenesis or hypogenesis of the corpus callosum frequently coexists with other anomalies. Thus when partial or complete corpus callosum agenesis is found to be present, it is important to look for other malformations of the brain. It is not unusual in these cases to find associated ▶ abnormalities of cortical development such as polymicrogyria, pachygyria, cortical dysplasia, or migration disorders. Malformations of the posterior cranial fossa may also occur. Foremost among these is ▶ Chiari malformation type II, which has a high association with corpus callosum agenesis or hypogenesis. The classic signs of Chiari malformation type II include downward displacement of the cerebellar tonsils, a “beaked” tectum, compression and flattening of the fourth ventricle and pons, and kinking of the medulla oblongata. Note When corpus callosum agenesis is present, always look for a possible coexisting Chiari malformation (and vice versa). Another relatively common disorder associated with corpus callosum agenesis is ▶ Dandy–Walker malformation, characterized by an enlarged posterior fossa with absence or hypoplasia of the cerebellar vermis and cystic dilatation of the fourth ventricle. Conversely, when this malformation is present, it is important to look for possible agenesis or hypogenesis of the corpus callosum. There are also many syndromes, some based on a chromosome abnormality, that are associated with agenesis or hypogenesis of the corpus callosum. They include: Aicardi’s syndrome. Apert’s syndrome. Cogan’s syndrome. Fetal alcohol syndrome. Morning glory syndrome. Rubinstein–Taybi syndrome. During embryonic and early fetal development, the brain is invested by a “primitive meninx” composed of still-undifferentiated mesenchyme with pluripotent cells. An intracranial lipoma forms when these cells undergo abnormal differentiation and develop into fat cells. Intracranial lipomas, then, are dysontogenetic tumors without neoplastic potential and generally do not require treatment. Intracranial lipomas are most commonly located in the interhemispheric fissure and are often associated with agenesis or hypogenesis of the corpus callosum. Lipomas at that location constitute typical midline deformities and, as such, may be associated with other midline defects such as cleft lip and palate. Less common sites of occurrence are the cerebellopontine angle and the suprasellar or supracerebellar cisterns. MRI findings Intracranial lipomas are isointense to fat in all sequences. A sequence with a fat-saturation pulse or in-phase and opposed-phase sequences, which lead to a chemical shift artifact, are sometimes helpful in making a diagnosis. As a rule, however, lipomas are easy to diagnose in conventional sequences. On the one hand, they are associated with the classic signs of corpus callosum agenesis described above. On the other, T1w sequences demonstrate a markedly hyperintense structure, isointense to fat, located within the interhemispheric fissure. Vessels in the interhemispheric fissure are not displaced by the mass—instead, they typically pass through the lipoma, which simply consists of abnormally differentiated tissue ( ▶ Fig. 9.17). Intracranial lipomas may calcify over time, causing decreased signal intensity in T2w sequences. Calcified lipomas are occasionally detected on skull radiographs. Fig. 9.17 Agenesis of the corpus callosum with a midline lipoma. The images display a midline mass isointense to fat and partial agenesis of the corpus callosum. Peripheral calcification of the lipoma is also apparent. (a) Sagittal T2w image. (b) Sagittal T1w image. (c) Coronal T2w image. Agenesis of the corpus callosum is often associated with cysts of the interhemispheric fissure, producing a characteristic malformation pattern. Interhemispheric cysts are divided into two main types: Type 1 interhemispheric cysts, which communicate with the ventricular system. Type 2 interhemispheric cysts, which do not communicate with the ventricular system. Clinical manifestations and epidemiology The clinical presentation of interhemispheric cysts varies with the severity of the malformation. Agenesis or hypogenesis of the corpus callosum with interhemispheric cysts may be almost clinically silent and is occasionally noted as an incidental finding. Some patients, however, may experience refractory epileptic seizures and developmental abnormalities. The incidence of congenital interhemispheric cysts is slightly higher in males than females. MRI findings Type 1 interhemispheric cysts MRI in these cases demonstrates a unilocular midline cyst, usually relatively large, that is isointense to CSF. Type 1a interhemispheric cysts communicate with the lateral ventricles, while type 1b cysts communicate with the third ventricle. Both of these subtypes are associated with macrocephaly. Type 1c interhemispheric cysts, on the other hand, are associated with microcephaly; the cyst communicates with the lateral ventricles and third ventricle. Type 1 interhemispheric cysts These cysts are usually multilocular ( ▶ Fig. 9.18) and do not communicate with the ventricular system. Type 2a interhemispheric cysts are isointense to CSF and cause macrocephaly. Type 2b interhemispheric cysts are hyperintense in T1w sequences and hyperdense on CT. They are often associated with subependymal heterotopias, polymicrogyria, and a defect in the falx cerebri. Type 2c interhemispheric cysts are isointense to CSF and often associated with focal subcortical heterotopias. Type 2d interhemispheric cysts are arachnoid cysts that are isointense to CSF and have no internal septa on MRI. Fig. 9.18 Interhemispheric cyst. T2w single-shot images of a fetus in week 32 show a large cyst with internal septa located in the interhemispheric fissure.
9.2 Abnormalities of Cortical Development
9.2.1 Group I Malformations
9.2.1.1 Microcephaly and Microcephaly with a Simplified Gyral Pattern
9.2.1.2 Megalencephalies (Group I.B) and Hemimegalencephalies
Megalencephalies

Hemimegalencephaly

9.2.1.3 Focal Cortical Dysplasias Type II (Group I.C)


9.2.2 Group II Malformations
9.2.2.1 Periventricular (Subependymal) Heterotopias (Group II.A) and Focal Subcortical Heterotopias (Group II.C)
Periventricular (Subependymal) Heterotopias

Focal Subcortical Heterotopias

9.2.2.2 Lissencephalies (Group II.B)
Classic Lissencephalies

Laminar Heterotopias

9.2.2.3 Cobblestone Malformations (Group II.D)
9.2.3 Group III Disorders
9.2.3.1 Polymicrogyria and Schizencephaly (Group III.A) and Polymicrogyria without Schizencephaly (Group III.B)
Polymicrogyria

Schizencephaly


9.2.3.2 Focal Cortical Dysplasia Types I and III (Group III.C)
Focal Cortical Dysplasia Type I

Focal Cortical Dysplasia Type III
9.3 Malformations of the Corpus Callosum and Commissures



9.3.1 Malformations and Syndromes Associated with Agenesis of the Corpus Callosum
9.3.2 Intracranial Lipomas with Corpus Callosum Agenesis

9.3.3 Interhemispheric Cysts with Corpus Callosum Agenesis

Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


