Location: In association with major nerve trunks: sciatic nerve, brachial plexus, sacral plexus. Gluteal region and pelvis, thigh region, shoulder region, and axilla are the most frequent sites.
Clinical: Rapidly enlarging mass with pain. Sensory and motor symptoms including projected pain, paresthesias, and weakness.
Imaging: Neither CT nor MRI can establish a definite diagnosis. On CT: mild enhancement with some nonenhancing areas of necrosis. On MRI: inhomogeneous and higher intensity than muscle on T1, markedly inhomogeneous and bright on T2, common necrosis in the center of the mass with peripheral capsule-like or irregular enhancement on contrast T1, a wide halo of edema unusual in benign lesions.
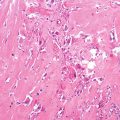
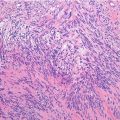
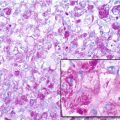
Histopathology: Large fusiform or eccentric mass within a major nerve. When the tumor spreads along the epi- and perineurium, it has a rosary bead aspect proximal or distal to the principal mass. Tumor is usually >5 cm, deep-seated, but rarely superficial, and fleshy, with hemorrhagic and necrotic diffused areas. Spindle cells with markedly irregular contours with wavy-, buckled-, or comma-shaped nuclei, lightly stained and usually indistinct cytoplasm. Densely cellular sweeping fascicules alternate with hypocellular, myxoid zones where the parallel orientation of cells is lacking. A peculiar nodular or whorled arrangement of cells can be present. Focal nuclear palisading is present in only 10 % of cases. Hemangiopericytoma-like vascular pattern can be present. Heterologous elements such as glandular differentiation, skeletal muscle (malignant triton tumor), bone, cartilage, and angiosarcomatous areas are present in about 15 % of cases. On immunohistochemistry, focal S-100 protein expression in spindled tumors; diffused strong S-100 protein expression in epithelioid MPNST.
Course and Staging: Frequent local recurrence when surgery is inadequate. Metastases frequently develop in the lungs, liver, subcutaneous, and bone, rarely in the lymph nodes. Usually, stage IIB.
Treatment: Very wide or radical local excision. Often, multicentric origin and diffusion along the nerve make even radical surgery ineffective. Overall 5-year survival is 30 % in patients with neurofibromatosis, 75 % in sporadic cases. In neurofibromatosis, prognosis is worse because the tumor involves the trunk and proximal extremities with late diagnosis, it is larger and of higher grade, and multiple sarcomas may occur. Usefulness of radiotherapy and chemotherapy is uncertain.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


