Fig. 7.1
Three-dimensional ultrasound with post-processing rendering using HDLive™ shows a midtrimester fetus with micrognathia and low-set ears (Courtesy of Dr. G. Grisolia)
After the sixth gestational week, mandibular development begins with fusion of ectoderm of the branchial arches and neural crest cells from the dorsal neural tube [3]. This complex process is influenced by both environmental and genetic factors. Micrognathia is the result of hypoplasia of the neural crest cell population and can occur in isolation as in primary mandibular disorders or, more frquently, associated with other conditions such as chromosomal aberrations (particularly triploidy, trisomy 18, and trisomy 9) (Fig. 7.2), genetic syndromes, and other structural anomalies (Fig. 7.3) [4, 5].
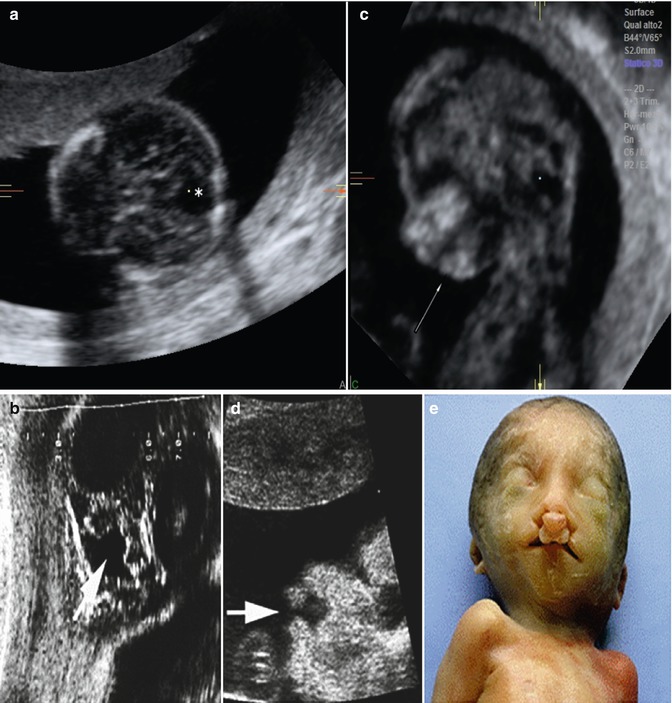
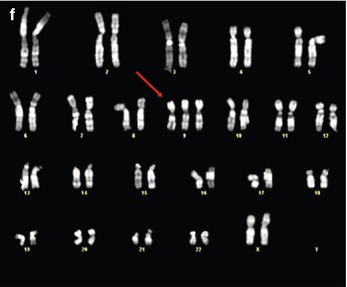
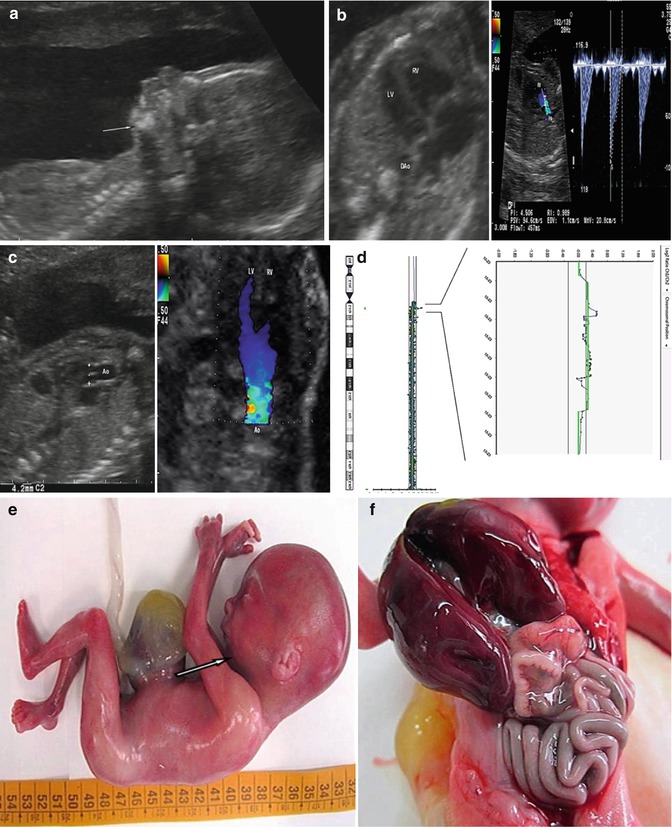
Fig. 7.2
Midtrimester fetus with (a, b) cerebellar vermian defect (arrow), (c) micrognathia (arrow), and (d) cleft lip and palate (arrow). (e) Postmortem examination confirmed bilateral cleft lip and palate. (f) This infant had trisomy 9 (From Tonni et al. [75])
Fig. 7.3
Micrognathia detected by two-dimensional ultrasound in a midtrimester fetus with normal karyotype (a) with associated tetralogy of Fallot (b, c) and exomphalos. Array-CGH demonstrated a 15q.11 microduplication (d), with the father as a carrier. Autopsy findings (e, f) confirm micrognathia (arrow) and exomphalos (From Tonni et al. [76])
Using the technology available in the current era of prenatal diagnosis, three-dimensional (3D) and four-dimensional (4D) ultrasound can visualize the mandible as early as 10 weeks of gestation and permit its detailed evaluation using indices, ratios, and facial angles, as well as mandibular growth charts [6, 7]. However, the diagnosis of a mandibular abnormality and its relationship with multiple malformations when present in complex syndromes remains challenging.
Although ultrasound is the established standard for fetal bone visualization, magnetic resonance imaging (MRI) may be a useful adjunct after 18 weeks of gestation by potentially providing additional details of the mandibular anatomy as well as of other abnormalities that may be present [8]. Despite these tools, assessing the severity of micrognathia is still a challenge for perinatologists. Nevertheless, prenatal evaluation of the fetal facial anatomy is necessary to optimize intrapartum management.
7.2 Genetic Syndromes Associated with Micrognathia
7.2.1 Pierre Robin Syndrome
Pierre Robin Syndrome (PRS, OMIM 261800) is a rare heterogeneous genetic disorder characterized by retro- or micrognathia, glossoptosis, and cleft palate independent of the respiratory and feeding difficulties or the size of the posterior cleft [9]. PRS is an autosomal recessive disorder caused by a mutation on chromosome 17q24.3-q25.1. In a recent study, the visualization of a cleft in the posterior palate in addition to retro- or micrognathia had a positive predictive value of 100 % for PRS [10]. The incidence of PRS has been estimated to occur in 1 of 14,000 live births [11]. PRS can be associated with several genetic disorders including trisomy 21, trisomy 13 [12], 46,XX/XY del(22)(q11), del(4)(q31), del(4)(p-), inv(9)(p11;q12), inv(9)(p11q13), t(X;2) and del(14(qter) [9]. In addition to the classic triad (retro- or micrognathia, glossoptosis, and cleft palate), PRS is also associated with other malformations such as macro- or microglossia, auricular malformations, nasal deformities, dental malformations, and laryngomalacia. Extracranial anomalies are observed in 10–85 % of cases and consist of ocular, cardiovascular, musculoskeletal, neurologic, and genitourinary abnormalities [13].
Prenatal diagnosis of PRS is critical because the tongue may significantly obstruct the airway at the time of birth [2]. Antenatal identification, therefore, allows delivery planning with a multidisciplinary team present at delivery to assist the newborn [12]. The diagnosis is most commonly made in the second trimester of pregnancy when severe retro- or micrognathia is visualized on two-dimensional (2D) ultrasound. Typically, polyhydramnios subsequently develops due to obstruction of oropharynx [13]. 3D ultrasound in the rendering mode allows improved spatial visualization of the fetal face, which is useful for counseling of the parents (Figs. 7.4 and 7.5) [13]. First-trimester diagnosis of PRS has been reported after transvaginal evaluation identified the typical facial dysmorphisms [14, 15]. MRI may also be helpful when the diagnosis is uncertain [8, 16]. However, the detection rate of PRS using 2D ultrasound is low, ranging from 7 to 22 % [17].

Fig. 7.4
Micrognathia (arrow) detected by three-dimensional ultrasound in a second-trimester fetus (a) with trisomy 22 (b) (From Tonni et al. [77])
After PRS is prenatally identified, the parents should strongly consider genetic amniocentesis because many different genetic disorders may be associated with this syndrome. These include trisomy 13, trisomy 18, cerebrocostomandibular syndrome, CHARGE syndrome (coloboma, heart anomaly, choanal atresia, retardation, genital abnormalities, and ear anomalies), velocardiofacial syndrome, Treacher Collins syndrome, Beckwith–Wiedemann syndrome, Cornelia de Lange syndrome, Smith–Lemli–Opitz syndrome, and Hanhart syndrome [10, 14]. Several cases of PRS have also been reported in the context of Stickler syndrome, which is a rare autosomal dominant connective tissue disorder estimated to affect approximately 1 in 7500 newborns [18]. Stickler syndrome, however, is characterized by ocular manifestations, arthritic changes, orofacial features, and deafness. Ophthalmologic anomalies are the most characteristic and most serious manifestations of the syndrome [19]. The presence of PRS phenotype during prenatal ultrasound in a patient with a family history of Stickler syndrome should raise significant suspicion of recurrence.
The prognosis of PRS usually is very good when it is isolated. After birth, tracheostomy is usually indicated to improve respiratory function. Surgeries to correct the mandibular dysmorphology and the cleft palate are performed between 6 and 18 months of life. Breathing difficulties gradually improve and may resolve completely due to growth of the mandible [13]. The long-term outcome depends on whether the PRS is isolated or associated with other malformations or syndromes. Even when there are associated anomalies, the survival rate of PRS can be as high as 90 %, although mental retardation occurs in 15 % of cases [12].
7.2.2 Neu–Laxova Syndrome
Neu–Laxova syndrome (NLS, OMIM 256520) is a rare, autosomal recessive syndrome that is lethal. NLS is characterized by severe intrauterine growth restriction (IUGR), flexion contractures of the limbs, microcephaly, edema particularly affecting the hands and feet, and ichthyosis [20]. Additional features include micrognathia, exophthalmos, cystic hygroma, cleft lip/palate, lissencephaly, cryptorchidism, and pulmonary hypoplasia [21].
The prenatal diagnosis of NLS by 2D ultrasound is possible in the second and third trimesters of pregnancy. As a lethal condition, the appropriate prenatal diagnosis is important to the parental counseling, particularly for consanguineous parents [22]. The main prenatal ultrasound findings in a series of seven cases are the following: low-set/malformed ears, flat/abnormal nose, micrognathia, microcephaly, rocker-bottom feet, IUGR, short neck, and increased adipose tissue [20]. The absence of breathing movements, suckling, swallowing, or isolated arm and leg movements may also be detected in the third trimester [23]. The association of this syndrome with polyhydramnios is reported in the literature [21, 24]. MRI is important to better assess the central nervous system for abnormalities.
Differential diagnoses include fetal akinesia/hypokinesia syndrome, cerebro-oculo-facioskeletal (COFS) syndrome, lethal multiple pterygium syndrome, Pena–Shokeir phenotype, Cornelia de Lange syndrome, Freeman–Sheldon syndrome, Miller–Dieker syndrome, Seckel syndrome, Smith–Lemli–Opitz syndrome, and intrauterine infections [21, 24]. The karyotype is typically normal [24].
7.2.3 Wolf–Hirschhorn Syndrome
Wolf–Hirschhorn syndrome (WHS, OMIM 194190) is a congenital disorder caused by microdeletion of the short arm of chromosome 4 (del 4p16.3), which encodes genes including MSX1, WHSC1, and LETM1 [25]. Most of these deletions occur de novo, but 15 % are due to a structural chromosomal rearrangement in one of the parents [26]. The incidence of this rare condition has been estimated to be around 1/50,000 births [27]. WHS is characterized by intrauterine growth restriction (IUGR), mental retardation, characteristic facial dysmorphism, microcephaly, ear lobe anomalies, and closure defects (cleft lip or palate, coloboma of the eye, and cardiac septal defects) [26]. The prenatal diagnosis is possible in the second and third trimesters by 2D and 3D ultrasound when the following malformations are observed: severe IUGR, microcephaly, and a characteristic facies with “Greek warrior helmet” appearance (prominent glabella, high-arched eyebrows, hypertelorism, and a broad high nasal bridge continuing to the forehead), short philtrum, carp-shaped mouth, micrognathia, and low-set ears that frequently have pits and tags (Fig. 7.6) [28, 29]. In a series of 10 cases, the main prenatal ultrasound findings were IUGR, microcephaly, high forehead, micrognathia, prominent glabella, and hypertelorism [30]. Hypospadias in male fetuses is another prominent finding [29].
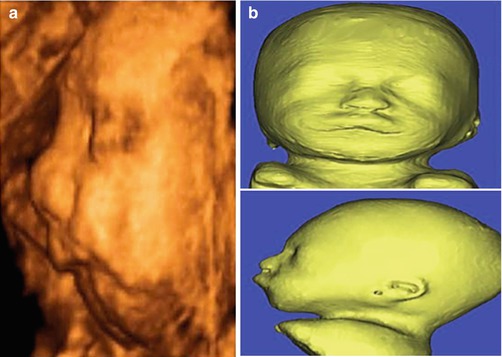
Fig. 7.5
(a) Three-dimensional ultrasound in surface rendering mode shows micrognathia in a fetus with Pierre Robin syndrome. (b) Three-dimensional virtual physical model printed on photopolymerized resin
The microdeletion of the short arm of chromosome 4 is a de novo mutation in most cases; parental testing may demonstrate a balanced translocation in other cases [31]. The main differential diagnoses are the following: Pitt–Rogers–Danks syndrome, Opitz BBB (oculogenitolaryngeal) syndrome, Fryns syndrome, Jacobsen syndrome, triploidy, trisomy 9, and trisomy 18 [25, 32].
The prognosis of WHS depends on its associated malformations. When congenital heart disease is present, there is a 30 % risk of mortality in first 2 years of life. Epilepsy is a primary characteristic; mental retardation is also common. Respiratory infections (including aspiration pneumonia, otitis media, sinusitis, or chronic cough) are very common finding in patients with WHS. Children with WHS also suffer from immunodeficiency. Other complications are the various tooth anomalies, including multiple tooth agenesis and taurodontism [25].
7.2.4 Cornelia de Lange Syndrome
Cornelia de Lange syndrome (CDLS, OMIM 122470), also known as Brachmann–de Lange syndrome [33], has an estimated prevalence of 1 in 10,000. Most cases are sporadic resulting from de novo dominant mutations [34]. Mutations of the NIPBL gene, which are typically de novo and associated with an autosomal dominant inheritance, are present in approximately 60 % of patients with CDLS [34]. The main characteristics in this syndrome are facial and limb abnormalities that present in combination with intrauterine and postnatal growth restriction [35]. Other frequent findings are cardiac defects, hirsutism, gastrointestinal abnormalities, and severe mental retardation [36].
Prenatal diagnosis of CDLS is possible by 2D and 3D ultrasound in the second and third trimesters of pregnancy, although the accuracy is low [37]. Prenatal ultrasound findings include severe IUGR <5th percentile, long eyelashes, disproportionate shortening of the long bones, depression of the bridge of the nose, and overriding of the upper lip (Fig. 7.7) [38–40]. In a report of seven cases, the main prenatal ultrasound findings were the following: IUGR, radial dysplasia/aplasia, ulnar dysplasia/aplasia, oligodactyly, small nose, prominent upper lip, micrognathia, and skin edema [41].
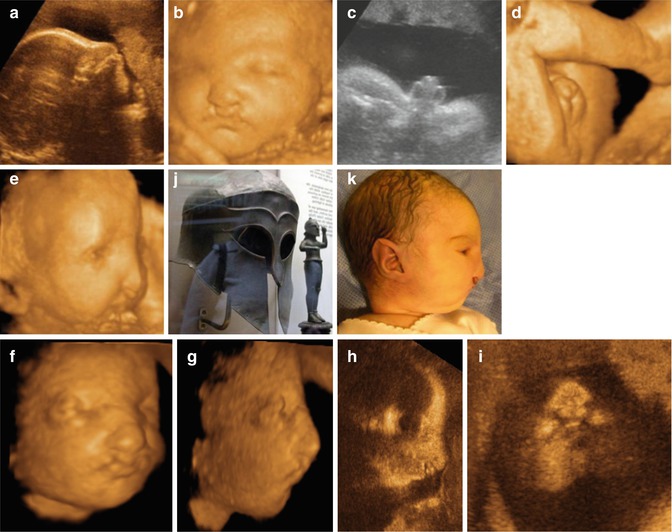
Fig. 7.6
Two- and three-dimensional findings in a male fetus with Wolf-Hirschhorn syndrome (a-i). Example of Greek warrior helmet (j) and infant with this resemblance (k) (From Sepulveda [29])
Although the syndrome is heterogeneous, the overall prognosis is rather poor as 20 % of affected children die during the first 2 years of life, while the rest usually face feeding problems, neurodevelopmental delay, and mental retardation similar to autism [42]. Children with CDLS have significant developmental delay and are unable to live without community support; even when phenotypic features are mild, children show poor communication, a delay in the acquisition of verbal skills, and overall significantly impaired cognitive development [43]. Because of this poor prognosis, prenatal diagnosis is very important to provide parental counseling and the option for termination of pregnancy.
7.2.5 Nager Syndrome
Nager syndrome (NS, OMIM 154400), or acrofacial dysostosis, is a heterogeneous syndrome that combines mandibulofacial dysostosis (micrognathia and ear anomalies) with limb defects [44]. The etiology is unknown; both autosomal dominant and recessive inheritance patterns have been hypothesized in some families, and teratogens such as retinoic acid have been linked [44, 45]. Heterozygous mutations in the SF3B4 gene on chromosome 1q12–q21 were found to be responsible for a subset of sporadic and autosomal dominant cases [46]. Depending on the type of limb defect, two major groups have been defined: NS, which has predominant preaxial anomalies, and Genee–Wiedemann or Miller syndrome, which is associated with postaxial malformations [47].
Few cases of NS that have been diagnosed in the prenatal period have been described in the literature. However, the diagnosis is possible in the second and third trimesters based on the following 2D and 3D ultrasound findings: severe micrognathia and limb malformations including the absence of thumbs, the absence of both the fibula and tibia, and shortening of the humerus and femurs (Fig. 7.8). Familiar cases of acrofacial dysostosis may help in the diagnosis. The karyotype is usually normal [44, 45, 47, 48].
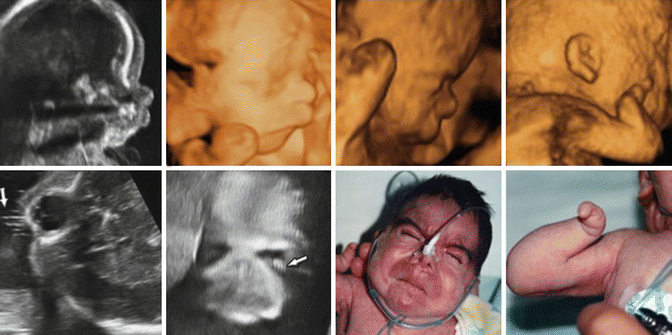
Fig. 7.7
Two- and three-dimensional ultrasound findings in fetus with Cornelia de Lange syndrome. Note frontonasal edema, micrognathia, long philtrum, long eyelashes, and abnormal upper limb (From Sepulveda et al. [39])
The differential diagnosis includes trisomy 18, Treacher Collins syndrome, Richieri–Costa–Pereira syndrome, thrombocytopenia–absent radius (TAR) syndrome, and Roberts syndrome [47]. Recently, SF3B4 mutations have been associated with NS; this suggests that at least some cases of Rodriguez syndrome are either allelic to or represent unusually severe manifestations of NS [49].
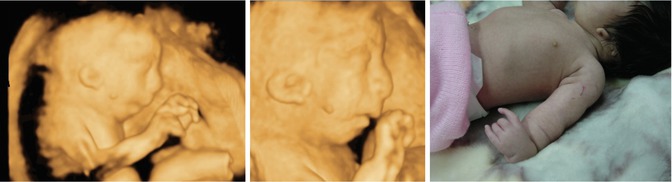
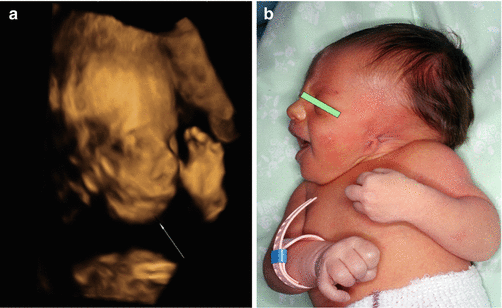
Fig. 7.8
Three-dimensional ultrasound in a fetus with Nager syndrome. Note micrognathia, abnormal ears, skin tag, and abnormal upper limbs
Fig. 7.9
(a) Three-dimensional ultrasound (3D) shows micrognathia (white arrow) in a fetus with Treacher Collins syndrome. (b) Photograph of the newborn infant shows micrognathia and abnormal ear
Prenatal diagnosis is important because the severe micrognathia can obstruct the upper airways with risk of respiratory insufficiency [48]. The prognosis is poor, and these children undergo several surgeries during childhood to correct the facial and limb dysmorphisms. Genetic counseling, including the option of termination of pregnancy, is important [47].
7.2.6 Femoral–Facial or Femoral Hypoplasia–Unusual Facies Syndrome
Femoral–facial syndrome (FFS, OMIM 134780) or femoral hypoplasia–unusual facies syndrome was first described in 1975 by Daentl et al. and is characterized by femoral hypoplasia (or aplasia) and specific facial dysmorphic features. It is usually described in association with other malformations such as renal, neural, skeletal, and genital anomalies [50, 51].
FFS occurs sporadically in females and seems to be associated with maternal diabetes or hyperglycemia, exposure to drugs (thalidomide), viral infections, radiation, focal ischemia or trauma, as well as severe fetal constraint secondary to oligohydramnios [52]. Some studies have suggested an autosomal dominant pattern of inheritance; however, there are no reports of chromosomal abnormalities in their carriers [53].
The facial malformations include micrognathia, low-set or dysplastic ears, cleft lip and palate, upslanting palpebral fissures, short nose with broad tip, long philtrum, and thin upper lip. Skeletal anomalies include sacral dysgenesis, talipes equinovarus, absent or hypomorphic tibia or fibula, humeroradial synostosis, syndactyly, and polydactyly. Renal defects, such as abnormal collecting urinary system, dysplasia or agenesis of kidneys, or multicystic kidneys, may be present. Associated genital anomalies include cryptorchidism, hypoplastic labia, and macrophallus. Central nervous system defects include ventriculomegaly, hydrocephalus, partial agenesis of the corpus callosum, and neuronal migration defects [54]. The subtrochanteric region of the femoral cartilage is also affected and causes shortening of the femoral proximal region. This is typically unilateral, with only 10–15 % of cases involving the legs bilaterally [55].
The prenatal diagnosis of FFS relies on a major defect of the skeletal or craniofacial region in addition to the femoral hypoplasia. The presence of a third malformation, such as unilateral renal agenesis or a spinal or central nervous system abnormality, can lead to the diagnosis [51]. 2D ultrasound can provide a complete evaluation, but 3D ultrasound images may enhance the detection of facial dysmorphisms [51]. MRI may assist in the identification of abnormalities that can lead to the prenatal diagnosis.
7.2.7 Larsen Syndrome
Larsen syndrome (LRS, OMIM 150250) was first described in 1950. The facial features first described were a prominent forehead, a depressed nasal bridge, and widely spaced eyes. Additional reports have presented other characteristics such as hydrocephalus, cleft palate, cardiac malformations, and spinal abnormalities [56]. Autosomal dominant and autosomal recessive patterns of inheritance have been proposed. Different patterns of inheritance could explain the clinical variability that is seen with this syndrome [57].
Larsen-like syndrome, a lethal variant of LRS, was reported by Chen et al. [58]. The characteristic features were tracheomalacia, multiple joint dislocations, pulmonary hypoplasia, collagen abnormalities, and nonimmune hydrops. Other studies have also shown abnormalities of collagen of joint capsules, cartilage matrix, and hyaline cartilage of the trachea [59]. The presence of bifid tongue and severe micrognathia can be associated and cause respiratory problems at birth. Autosomal recessive inheritance was proposed for this variant [60].
Related posts:
 Congenital Subcutaneous Mixed Venous-Lymphatic Orofacial Malformation Associated with Macroglossia: Prenatal Diagnosis with Ultrasound and Fetal MRI
Congenital Subcutaneous Mixed Venous-Lymphatic Orofacial Malformation Associated with Macroglossia: Prenatal Diagnosis with Ultrasound and Fetal MRI
 The Genetics of Facial Cleft
The Genetics of Facial Cleft
 Median Cleft Lip and Palate, Cutaneous Nasal Polyps, and Corpus Callosum Lipoma: A Case of Pai Syndrome Associated with Ventricular Septal Defects
Median Cleft Lip and Palate, Cutaneous Nasal Polyps, and Corpus Callosum Lipoma: A Case of Pai Syndrome Associated with Ventricular Septal Defects
 Acromelic Frontonasal Dysplasia (Median Cleft Face Syndrome)
Acromelic Frontonasal Dysplasia (Median Cleft Face Syndrome)
 Magnetic Resonance Imaging (MRI) in the Evaluation of the Fetal Face
Magnetic Resonance Imaging (MRI) in the Evaluation of the Fetal Face
 The Role of 2D/3D/4D Ultrasound in the Prenatal Assessment of Cleft Lip and Palate
The Role of 2D/3D/4D Ultrasound in the Prenatal Assessment of Cleft Lip and Palate
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


