Fig. 2.1
Drawing in an anterior oblique view of the late fetal face showing the contributions of the various facial processes. Green indicates the frontonasal process; yellow, the lateral nasal processes; purple, the medial nasal processes; orange, the maxillary processes; and blue, the mandibular processes (Courtesy of Som and Naidich, AJNR, 2014 [4])
Each of the clefts, apart from the midline clefts, can be unilateral or bilateral, and the cleft lip can be with or without CP with the left side being more frequently affected than the right.
Cleft palate, on the other hand, is the result of incomplete fusión of the palatal shelves and can involve the primary palate, the secondary palate, or both, and can be unilateral or bilateral. In some cases, almost the entire hard and soft palate are missing (Fig. 2.2).
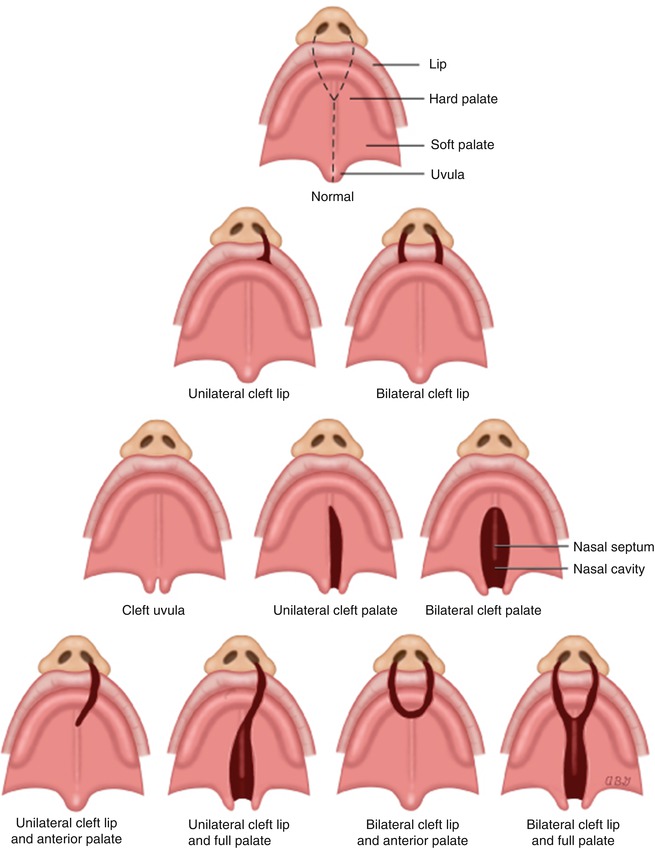
Fig. 2.2
Types of cleft lip ± palate
2.3 Non-syndromic Cleft Lip/Cleft Palate and Cleft Palate
Non-syndromic CL/P and CP have a multifactorial mode of inheritance. In this mode of inheritance, the defect is due to interaction between multiple genes, each having a small additive effect with environmental factors; when the combination of both components reaches a threshold, it results in the abnormality which can be of variable severity (Fig. 2.3). Of the genes involved, some have a major effect such as sonic hedgehog [5], transforming growth factor alpha and beta 3 [6, 7], and IRF6 [8], and some have small additive contributions such as polymorphisms for genes coding for enzymes in the folic acid metabolism pathway [9]. Of the environmental factors, most are not known but some have a major teratogenic effect when exposed during the first trimester of pregnancy. The most important teratogens associated with CL/P and CP are antiseizure medications (phenytoin, sodium valproate, and topiramate) [10] and folic acid antagonists such as methotrexate [11]. Other exposures suspected to be associated with CL/P and CP are cigarette smoking [12], folic acid deficiency [13], and exposure to corticosteroids [14].
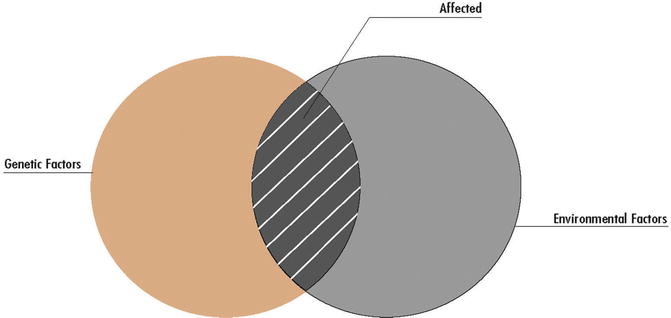
Fig. 2.3
Multifactorial mode of inheritance. Interaction between genes and environment (Courtesy of Greenwood Genetic Center)
As in other multifactorial conditions, the recurrence risk for isolated CL/P depends on the severity of the defect, the number of family members affected, gender (in CL/P, affected females have a higher recurrence risk), and ethnic background (American Indian/First Nations > Asian > Caucasian > African) (Table 2.1).
Relative | CL | CL/P | CP |
|---|---|---|---|
Sib | 2.5 | 3.9 | 3.3 |
Half sib | 1.0 | 0.5 | 1.0 |
Parent | 2.5 | 2.5 | 2.1 |
Offspring | 3.5 | 4.1 | 4.2 |
Niece/nephew | 0.9 | 0.8 | 1.1 |
Aunt/uncle | 0.6 | 1.1 | 0.6 |
First cousin | 0.3 | 0.5 | 0.4 |
The recurrence risk of isolated CP does not seem to be influenced by the ethnic background but is higher when a male is the proband.
2.4 Syndromic Cleft Lip/Palate and Cleft Palate
2.4.1 Chromosomal Abnormalities
Trisomy 13: This is also called Patau syndrome and has an incidence of about 1:16,000 newborns. In 80 % of the cases, the chromosome count is 47 and these cases are associated with maternal age. However, 20 % of the cases are due to translocation involving chromosome number 13, and some of these cases are inherited from a parent who carries a chromosome rearrangement involving chromosome number 13, usually as a Robertsonian translocation. The condition is associated with severe intellectual disability, facial dysmorphism, scalp defects, CL/P (occasionally midline) or CP, anophthalmia/microphthalmia, brain and cardiac abnormalities, genital malformations, and postaxial polydactyly. Only 5–10 % of the cases this condition live past their first year [16].
Trisomy 18: This is also called Edwards syndrome and has incidence of 1: 5,000 newborns. About 85 % of the cases recognized during pregnancy do not survive to term, and the chance of having a child with this condition increases with maternal age. Full trisomy 18 always occurs de novo and is associated with intrauterine growth restriction (IUGR) and low birth weight, severe intellectual disability, characteristic facial features, CL/P or CP, cardiac abnormalities, clenched fists with overlapping fingers, rocker-bottom feet, and prominent heels. Many individuals with trisomy 18 die in the first month of life, and only 5–10 % of the newborns live past their first year [16].
2.4.2 Microdeletion/Microduplication Syndromes
4p deletion: This is also known as Wolf-Hirschhorn syndrome and is associated with delayed growth and development, characteristic facial features (“Greek warrior helmet”), microcephaly, CL/P or CP, iris and/or optic nerve coloboma and other eye defects, scoliosis and kyphosis, club feet, and occasional cardiac and genital malformations. The prevalence of Wolf-Hirschhorn syndrome is estimated to be 1 in 50,000 births [17].
22q11.2 deletion syndrome: This is also known as velo-cardio-facial syndrome, DiGeorge syndrome, or Shprintzen syndrome, and is associated with a large interstitial deletion of 22q11.2. At least 30 genes have been mapped to the region, but most phenotypes of the syndrome are believed to be the haploinsufficiency of the gene TBX1. The condition is associated with short stature (1/3 of cases), CL/P or CP, conductive hearing loss usually secondary to the cleft palate, velopharyngeal incompetence, bulbous/square nose, narrow palpebral fissures, micro-/retrognathia, cardiac abnormalities, cellular immunodeficiency, and hyperparathyroidism (usually transient). Moderate or severe learning problems are present in ~20 %, and 10 % have psychiatric disorders (schizophrenia and/or bipolar disorder).
1q43–q44 deletion: This condition is characterized by prenatal and postnatal growth restriction, intellectual disability (moderate to severe), characteristic facial features including microcephaly, prominent forehead, short nose with a broad nasal tip, and CP. Agenesis of the corpus callosum and other brain abnormalities are common [18].
3q29 microdeletion syndrome: The condition is associated with variable phenotypes despite an almost identical deletion size. Common clinical findings include mild to moderate mental retardation, autism, gait ataxia and only slightly dysmorphic facial features including microcephaly, a narrow face, short philtrum, high nasal bridge, CP or CL/P, horseshoe kidney, hypospadias and ligamentous laxity [19].
2.5 Mendelian Syndromes Associated with Cleft Lip/Palate
2.5.1 Autosomal Dominant (AD)
van der Woude syndrome: This syndrome has an incidence of 1:35,000–1:100,000 and is caused by mutations in the interferon regulatory factor 6 (IRF6). It is characterized by lower lip pits (80 %), hypodontia, missing teeth, CL/P or CP, and cleft uvula. The average IQ of individuals with van der Woude syndrome is similar to that of the general population. Missense mutations in the same gene cause multiple pterygium syndrome [20].
Stickler syndrome: This AD disorder is characterized by Robin sequence, flat facies, myopia, retinal detachment and cataracts, spondyloepiphyseal dysplasia and CP and/or CL/P. Other findings include hearing loss (both sensorineural and conductive), hyperextensible joints, and talipes equinovarus. Gene mutations known to be associated with this condition occur in COL2A1, COL11A1, COL11A2, COL9A1, and COL9A2, which cause Stickler syndrome types I through V, respectively. These genes are involved in the production of three types of collagen: type II, type IX, and type XI. Stickler syndrome types I, II, and III are inherited in an autosomal dominant pattern, and types IV and V have an autosomal recessive mode of inheritance [21].
Treacher Collins syndrome: This condition is caused by mutations in the TCOF1 (78–93 %) and in the POLR1D and POLR1C genes. The dysmorphic features, although variable, are distinctive and consist of mandibular/malar hypoplasia, CP with or without cleft lip, hypoplastic zygomatic bone, lower eyelid coloboma, partial to total absence of the lower eyelashes, abnormal auricles/external ear canal, unilateral or bilateral choanal stenosis or atresia, and conductive deafness usually caused by malformation of the ossicles and hypoplasia of the middle ear cavities. Inner ear structures are usually normal. The incidence is about 1:50,000 [22].
Ectrodactyly–ectodermal dysplasia-clefting syndrome (EEC) or ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome: This condition is associated with CL/P (in about 2/3 of cases), maxillary and malar hypoplasia, short philtrum, ankyloblepharon, labyrinthine dysplasia and distal limb defects including syndactyly, camptodactyly, and ectrodactyly (in up to 85 % of cases). Ectodermal dysplasia includes sparse wiry hair, skin erosions, nail changes, dental changes, decreased sweating, and hypoplastic nipples. Partial anodontia and microdontia are common. Genitourinary anomalies are present in about half of the cases and include megaureter, duplicated collecting system, vesicoureteral reflux, ureterocele, bladder diverticula, and renal agenesis/dysplasia. The condition is caused by mutations in TP63 which is also known to be associated with Hay-Wells syndrome [23].Related posts:
 Congenital Subcutaneous Mixed Venous-Lymphatic Orofacial Malformation Associated with Macroglossia: Prenatal Diagnosis with Ultrasound and Fetal MRI
Congenital Subcutaneous Mixed Venous-Lymphatic Orofacial Malformation Associated with Macroglossia: Prenatal Diagnosis with Ultrasound and Fetal MRI
 Atypical Facial Cleft Detected by Prenatal Scan and Confirmed by Postmortem Reconstructive Computed Tomography (CT REC)
Atypical Facial Cleft Detected by Prenatal Scan and Confirmed by Postmortem Reconstructive Computed Tomography (CT REC)
 Acromelic Frontonasal Dysplasia (Median Cleft Face Syndrome)
Acromelic Frontonasal Dysplasia (Median Cleft Face Syndrome)
 The Fetal Brain in Fetuses with Orofacial Abnormalities
The Fetal Brain in Fetuses with Orofacial Abnormalities
 Magnetic Resonance Imaging (MRI) in the Evaluation of the Fetal Face
Magnetic Resonance Imaging (MRI) in the Evaluation of the Fetal Face
 The Role of 2D/3D/4D Ultrasound in the Prenatal Assessment of Cleft Lip and Palate
The Role of 2D/3D/4D Ultrasound in the Prenatal Assessment of Cleft Lip and Palate

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


