Fig. 9.1
Stillbirth with frontonasal dysplasia. Note the broad nasal cleft and extreme hypertelorism (a) and the presence of an occipital cephalocele (b)
The association between open neural tube defects and facial anomalies appears to be sporadic and not related to any known syndrome. In a population-based study, Stoll et al. reported on 156 cases of anencephaly/spina bifida diagnosed in Northern France and examined by a clinical dysmorphology geneticist; they found that 3.3 % suffered from facial clefts. Facial cleft was the most common associated anomaly but they were not syndromic [4]. Upon review of current literature, there was only one case that was associated with a de novo -2q deletion [5].
Anterior cephalocele has been described in fetuses with frontonasal dysplasia (FND), also known as median cleft facial syndrome (http://omim.org/entry/136760) [6, 7] (Table 9.1). This condition is usually sporadic, but autosomal dominant, recessive, and X-linked patterns, as well as 22q11 microdeletion, have been reported. Patients with FND usually present with two or more of the following: pronounced ocular hypertelorism, broadening of the nasal root, median facial cleft affecting the nose and/or upper lip and palate, unilateral or bilateral clefting of the alae nasi, lack of formation of the nasal tip, anterior cranium bifidum occultum, and a V-shaped or widow’s peak frontal hairline (Fig. 9.1). The corpus callosum (CC) and other important brain structures may be absent. Severe forms of FND, particularly when involving the brain, may be associated with autosomal recessive mutations in aristaless-like homeobox gene ALX1; ALX1 expression is essential for building oral and nasal cavities as well as proper eye development during early embryogenesis [8].
Table 9.1
Neural tube defects in fetuses with orofacial syndromes and anomalies
Neural tube defect | Facial anomalies | Syndrome | Other CNS findings | Genetics | References |
|---|---|---|---|---|---|
Anencephaly/spina bifida | Facial clefts | No | Sporadic, de novo deletion 2q | ||
Anterior cephalocele | Median cleft lip, hypertelorism | Frontonasal dysplasia | ACC, lipoma, macrocephaly, HME, cranium bifidum | Sporadic, ALX AR mutations, AD, X-linked patterns, 22q11 microdeletions | |
Posterior encephalocele | Microophthalmia/anophthalmia, orofacial clefts | Meckel–Gruber syndrome, other ciliopathies | Cervical rachischisis, DWM, ACC, hypotelorism, cerebellar hypoplasia, hydrocephalus | MKS1-11 AR sequence variations |
The Meckel–Gruber syndrome is the most severe form of the autosomal recessive ciliopathies and is characterized by the presence of posterior cephaloceles, polycystic kidneys, and polydactyly; it is also frequently accompanied by facial clefts (25 %) and microphthalmia/anophthalmia (20 %) [9] (Fig. 9.2). Less severe ciliopathies including Joubert’s syndrome and oral–facial–digital syndrome type IV may also be associated to a lesser extent with facial dysmorphism, cleft lip and palate, and facial tumors [10].
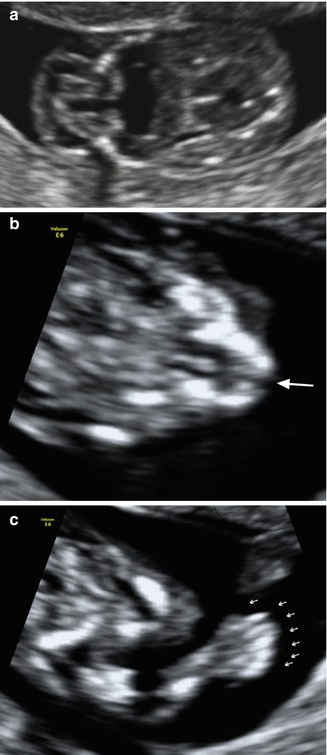
Fig. 9.2
Fetus with Meckel–Gruber syndrome identified at 12 weeks of gestation as demonstrated by TVS. (a) Occipital encephalocele; (b) note the presence of cleft lip (arrow) and (c) polydactyly
9.2 Disorders of Prosencephalic Development
Prosencephalic development starts as the same time as facial development at 5–6 weeks of gestation. At this stage, the brain is composed of three vesicles: the forebrain, the midbrain, and the hindbrain. Failures in the normal development of the forebrain result in a wide range of anomalies ranging from complete failure of formation to abnormal formation of some of the midline brain structures [11]. Failure of formation of the prosencephalon leads to the development of two very rare and severe conditions: aprosencephaly and atelencephaly; failure of cleavage into two separated vesicles results in holoprosencephaly, and anomalous midline development causes commissural and septal anomalies [1] (Tables 9.1 and 9.2).
Table 9.2
Classification of holoprosencephaly
HPE type | HPE subtype | Eye | Nose | Mouth | |
|---|---|---|---|---|---|
Classical forms | Alobar | Range from single orbit/eye to mild hypotelorism | Arrhinia, proboscis, single nostril | Agnathia, cleft lip and palate | |
Semilobar | Range from single orbit/eye to mild hypotelorism | Arrhinia, proboscis, single nostril | Agnathia, cleft lip and palate | ||
Lobar | Small OOD/IOD/normal | Normal | Cleft lip/palate | ||
Atypical forms | Middle interhemispheric variant | Normal | Normal | Normal | |
Septopreoptic | Normal | Normal | Single incisor | ||
Microforms | |||||
Isolated hypotelorism | Small OOD/IOD | Normal | Normal | ||
Single incisor | Normal | Normal | Normal |
Although relatively rare in live-born children (prevalence of less than 1/10,000), HPE is the most common fetal malformation, present in as many as 1 in 250 first-trimester fetuses with the more severe cases resulting in early fetal demise. HPE is associated with chromosomal anomalies, particularly trisomies 13 and 18, triploidy, other known syndromes, as well as single gene disorders.
With the advancements in molecular biology, an increasing number of single gene mutations have been described as causative agents of HPE; these mutations appear to occur with a wide range of different HPE genotypes confirming that HPE apparently represents a continuum of forebrain and craniofacial malformations with no clear-cut distinction among the different subcategories [12]. To date, four major genes responsible for HPE have been described: SHH, ZIC2, SIX3, and TGIF, and a few others seem to play a lesser role in the occurrence of HPE. The “multiple-hit hypothesis” is now the most widely accepted model of HPE. According to this hypothesis, combinations of mutations in major and/or minor HPE genes lead to the occurrence of HPE and may account for variability in terms of severity [13].
In a large European study [14] consisting of 645 probands, of which 51% were fetuses, and 699 relatives systematic molecular analyses showed point mutations or deletions in the four major genes involved in HPE (SHH, ZIC2, SIX3, TGIF) in 25.4 % of the subjects. More fetuses than children were found to carry an HPE-associated gene deletion (68 % vs. 32 %). CGH analysis performed on 260 of the probands demonstrated rearrangements in 22% of cases. Interestingly, HPE spectrum disorders were found in 79 of the 699 relatives (29 % in parents and 71 % in other relatives). All affected parents were diagnosed with brain microforms, which accounted for 44 % of the abnormalities in these 79 relatives. More severe forms were reported in other relatives, such as siblings or cousins: 26 % alobar HPE, 13 % semilobar HPE, and 7 % lobar HPE. Associated brain malformations were observed in 6 % of the affected relatives, and neural tube defects were found in 8 %.
An association between the severity of facial anomalies and brain anomalies (as in the maxim, “the face predicts the brain”) was first reported by DeMyer and colleagues in 1964 [15]. Since then, however, as experience with anomalies has increased, this “face-brain” correlation has become controversial [16]. In a large epidemiologic study, Orioli found that in 10–39 % of the cases, there was no such clear correlation between face and brain anomaly subtypes [17]. In contrast, Mercier et al. [14], based on the data for 369 informative probands, found a positive correlation between the severity of type of HPE and severity of facial features for their entires series (p < 0.001) and also for patients with SHH (p < 0.001), SIX3 (p < 0.001), and TGIF (p < 0.001) subgroups, but not for the ZIC2 subgroup (p > 0.5).
Prenatal diagnosis of the alobar and semilobar forms of HPE is usually straightforward; the first reported cases in the early 1980s were diagnosed in the second half of pregnancy and represented severe HPE types [18] (Figs. 9.3 and 9.4). In recent years, diagnosis of these severe cases is possible at much early gestational ages, even as early as 10–14 weeks [19, 20] (Fig. 9.5). Sepulveda and Wong demonstrated that failure to visualize the choroid plexuses, the “butterfly” sign, during the first trimester is highly predictive of the presence of HPE, and they were able to diagnose 11 cases out of 11,068 fetuses scanned without false-positive or false-negative cases [21]. In alobar and semilobar holoprosencephaly, the first clue for diagnosis may be either the visualization of an abnormal brain or an abnormal face. CNS findings include the presence of a large single ventricle with a thin cerebral mantle, failure to visualize the choroid plexuses, or the visualization of a large noncleaved thalamus (Figs. 9.3, 9.4, 9.5). It may be extremely difficult to differentiate between a fetus with HPE and one with severe hydrocephaly and ruptured septum pellucidum [22]. In these cases, as well as when lobar HPE is suspected, the best way to reach a conclusive diagnosis is by visualization of the most rostral portions of the cerebral hemispheres and frontal horns of the lateral ventricles in a coronal plane; all three classical forms of HPE will have a common frontal ventricle and at least some degree of non-cleavage of the frontal lobe without a normal interhemispheric fissure (Fig. 9.6). Visualization of two separated frontal horns at their foremost anterior position confidenty rules out a classic form of HPE. In these cases, other important findings are the presence of an abnormal anterior cerebral artery trajectory running closer than usual to the anterior subarachnoid space, a single choroid plexus, and the presence of craniofacial anomalies.
 Congenital Subcutaneous Mixed Venous-Lymphatic Orofacial Malformation Associated with Macroglossia: Prenatal Diagnosis with Ultrasound and Fetal MRI
Congenital Subcutaneous Mixed Venous-Lymphatic Orofacial Malformation Associated with Macroglossia: Prenatal Diagnosis with Ultrasound and Fetal MRI
 The Genetics of Facial Cleft
The Genetics of Facial Cleft
 Median Cleft Lip and Palate, Cutaneous Nasal Polyps, and Corpus Callosum Lipoma: A Case of Pai Syndrome Associated with Ventricular Septal Defects
Median Cleft Lip and Palate, Cutaneous Nasal Polyps, and Corpus Callosum Lipoma: A Case of Pai Syndrome Associated with Ventricular Septal Defects
 Acromelic Frontonasal Dysplasia (Median Cleft Face Syndrome)
Acromelic Frontonasal Dysplasia (Median Cleft Face Syndrome)
 Magnetic Resonance Imaging (MRI) in the Evaluation of the Fetal Face
Magnetic Resonance Imaging (MRI) in the Evaluation of the Fetal Face
 The Role of 2D/3D/4D Ultrasound in the Prenatal Assessment of Cleft Lip and Palate
The Role of 2D/3D/4D Ultrasound in the Prenatal Assessment of Cleft Lip and Palate




Related posts:
Congenital Subcutaneous Mixed Venous-Lymphatic Orofacial Malformation Associated with Macroglossia: Prenatal Diagnosis with Ultrasound and Fetal MRI
The Genetics of Facial Cleft
Median Cleft Lip and Palate, Cutaneous Nasal Polyps, and Corpus Callosum Lipoma: A Case of Pai Syndrome Associated with Ventricular Septal Defects
Acromelic Frontonasal Dysplasia (Median Cleft Face Syndrome)
Magnetic Resonance Imaging (MRI) in the Evaluation of the Fetal Face
The Role of 2D/3D/4D Ultrasound in the Prenatal Assessment of Cleft Lip and Palate
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
