of articular cartilage (Fig. 30.6), and hypertrophy of the soft tissues may also occur, leading to the development of square, spade-shaped fingers.
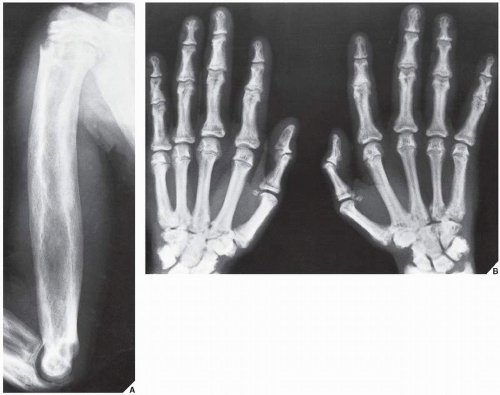 FIGURE 30.1 Familial idiopathic hyperphosphatasia. (A) Anteroposterior radiograph of the shoulder and arm of a 12-year-old Puerto Rican boy reveals marked thickening of the cortex of the humerus and coarsening of the bony trabeculae, resembling pagetic bone. (B) Radiograph of the hands shows sclerotic changes in the bones and a marked narrowing of the medullary cavity of the metacarpals and phalanges. |
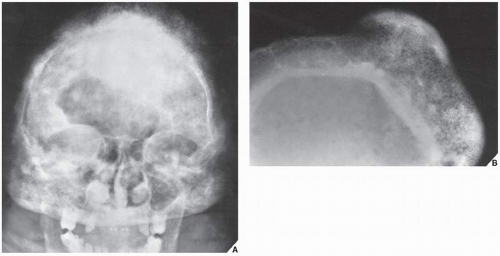 FIGURE 30.2 Familial idiopathic hyperphosphatasia. (A) Anteroposterior radiograph of the skull of a 30-year-old man shows calvarial thickening and sclerosis resembling that of Paget disease. (B) Magnification study reveals marked thickening of the inner table and widening of the diploë. |
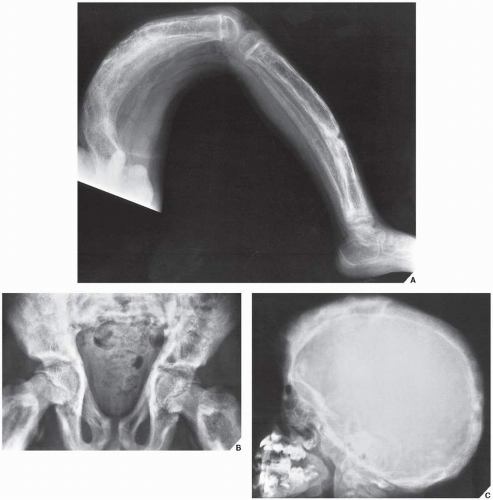 FIGURE 30.3 Familial idiopathic hyperphosphatasia. (A) Radiograph of a 4-year-old boy demonstrates marked bowing of the long bones of the lower extremity, a striking feature of this disorder. (B) Anteroposterior radiograph of the pelvis shows the coarse trabecular pattern and cortical thickening typical of this condition. Note that the epiphyses are not affected. (C) Lateral radiograph of the skull demonstrates thickening of the tables and a “cotton ball” appearance of the cranial vault, similar to that of Paget disease. (B from Sissons HA, Greenspan A, 1986, with permission.) |
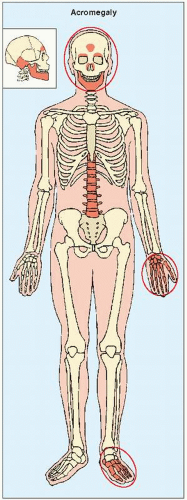 FIGURE 30.4 The most clearly revealing target sites of acromegaly. |
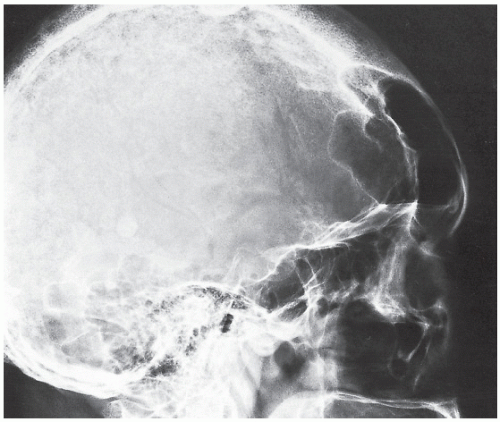 FIGURE 30.5 Acromegalic skull. Lateral radiograph of the skull of a 75-year-old woman shows marked enlargement of the frontal sinuses, prominent supraorbital ridges, and thickening of the frontal bones. |
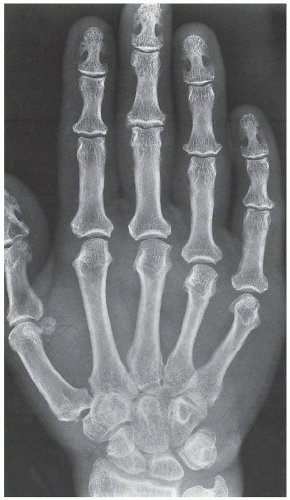 FIGURE 30.6 Acromegalic hand. Dorsovolar radiograph of the hand of a 38-year-old woman shows characteristic overgrowth of the terminal tufts and spur-like projections. The bases of the terminal phalanges are also enlarged, and the radiographic joint spaces are widened. |
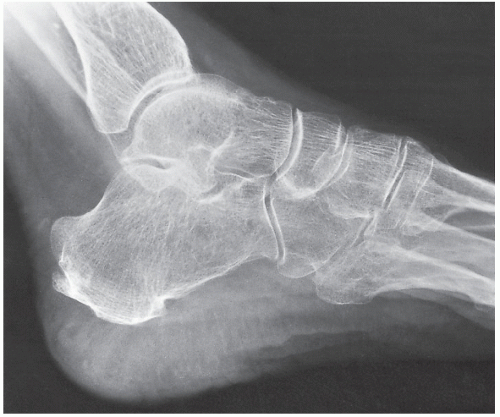 FIGURE 30.7 Acromegalic foot. Lateral radiograph of the foot of a 58-year-old man shows a heel-pad thickness of 38 mm, far above normal for this patient who weighs only 140 lb. This measurement corresponds to the shortest distance between the calcaneus and the plantar aspect of the heel. |
TABLE 30.1 Causes of Scalloping in Vertebral Bodies | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
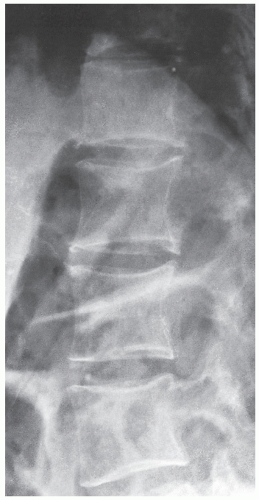 FIGURE 30.8 Acromegalic spine. Lateral radiograph of the thoracolumbar spine of a 49-year-old woman demonstrates posterior vertebral scalloping, a phenomenon apparently caused by bone resorption. |
Type I: | The nonneuronopathic, or adult type, is the most common form, occurring mainly in Ashkenazi Jews. Onset is in the patient’s first or second decade, and the individuals affected usually live normal life spans. Bone abnormalities and hepatosplenomegaly characterize this form of the disease, although some patients may not show any symptoms. |
Type II: | The acute neuronopathic form is lethal within the patient’s 1st year. This type apparently has no predilection for any ethnic group. Hepatosplenomegaly is invariably present, in addition to brain damage and seizure disorder. |
Type III: | The subacute juvenile neuronopathic form, occuring mainly in Swedish nationality from the Norbotten region begins in the latter part of the 1st year and follows a malignant course similar to that of type II. Patients present with hepatosplenomegaly, anemia, respiratory problems, mental retardation, and seizures, and usually die by the end of their second decade of life. |
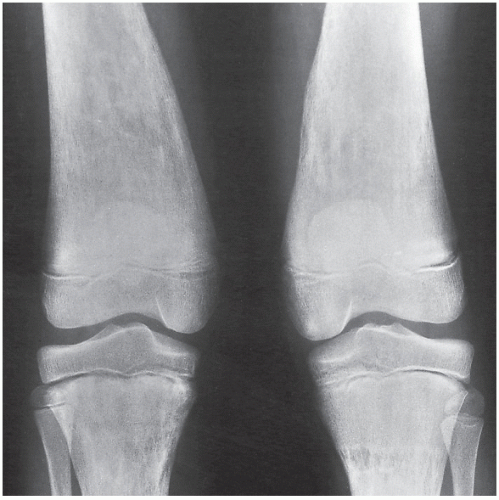 FIGURE 30.9 Gaucher disease. Anteroposterior radiograph of a 12-year-old boy with adult-type of disease shows the Erlenmeyer-flask deformity of both distal femora, secondary to medullary expansion. Note the thinning of the cortex caused by diffuse osteoporosis. |
to a bone-within-bone phenomenon, which may resemble osteomyelitis (Fig. 30.12). Recently, Hermann and associates conducted a study of 29 patients with type I Gaucher disease using MRI to determine the usefulness of this technique in the evaluation of bone marrow involvement. The results of this investigation suggest that MRI is a valuable noninvasive modality in this respect to assess disease activity. Apparently, the patients with decreased signal intensity within bone marrow on both T1-weighted and T2-weighted images, but showing a relative increase in signal intensity from T1 weighting to T2 weighting can be considered to have an “active process” that correlates well with their symptoms.
Related posts:
 Radiologic Evaluation of Skeletal Anomalies
Radiologic Evaluation of Skeletal Anomalies
 Benign Tumors and Tumor-like Lesions III: Fibrous, Fibroosseus, and Fibrohistiocytic Lesions
Benign Tumors and Tumor-like Lesions III: Fibrous, Fibroosseus, and Fibrohistiocytic Lesions
 Upper Limb III: Distal Forearm, Wrist, and Hand
Upper Limb III: Distal Forearm, Wrist, and Hand
 Benign Tumors and Tumor-Like Lesions IV: Miscellaneous Lesions
Benign Tumors and Tumor-Like Lesions IV: Miscellaneous Lesions
 Upper Limb III: Distal Forearm, Wrist, and Hand
Upper Limb III: Distal Forearm, Wrist, and Hand
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


