Molecular Mechanisms of DNA and Chromosome Damage and Repair
▪ GENERAL OVERVIEW OF DNA STRAND BREAKS
There is strong evidence that DNA is the principal target for the biologic effects of radiation, including cell killing, carcinogenesis, and mutation. A consideration of the biologic effects of radiation, therefore, begins logically with a description of the breaks in DNA caused by charged-particle tracks and by the chemical species produced.
Deoxyribonucleic acid (DNA) is a large molecule with a well-known double helical structure. It consists of two strands held together by hydrogen bonds between the bases. The “backbone” of each strand consists of alternating sugar and phosphate groups. The sugar involved is deoxyribose. Attached to this backbone are four bases, the sequence of which specifies the genetic code. Two of the bases are single-ring groups (pyrimidines); these are thymine and cytosine. Two of the bases are double-ring groups (purines); these are adenine and guanine. The structure of a single strand of DNA is illustrated in Figure 2.1. The bases on opposite strands must be complementary; adenine pairs with thymine, and guanine pairs with cytosine. This is illustrated in the simplified model of DNA in Figure 2.2A.
Radiation induces a large number of lesions in DNA, most of which are repaired successfully by the cell and are discussed in the following sections of this chapter. A dose of radiation that induces an average of one lethal event per cell leaves 37% still viable; this is called the D0 dose and is discussed further in Chapter 3. For mammalian cells, the x-ray D0 usually lies between 1 and 2 Gy. The number of DNA lesions per cell detected immediately after such a dose is approximately:
Base damage, >1,000 Single-strand breaks (SSBs), 1,000 Double-strand breaks (DSBs), 40
If cells are irradiated with a modest dose of x-rays, many breaks of a single strand occur. These can be observed and scored as a function of dose if the DNA is denatured and the supporting structure is stripped away. In intact DNA, however, SSBs are of little biologic consequence as far as cell killing is concerned because they are repaired readily using the opposite strand as a template (Fig. 2.2B). If the repair is incorrect (misrepair), it may result in a mutation. If both strands of the DNA are broken and the breaks are well separated (Fig. 2.2C), repair again occurs readily because the two breaks are handled separately.
By contrast, if the breaks in the two strands are opposite one another or separated by only a few base pairs (Fig. 2.2D), this may lead to a DSB (double-strand break), resulting in the cleavage of chromatin into two pieces. DSBs are believed to be the most important lesions produced in chromosomes by radiation; as described
in the next section, the interaction of two DSBs may result in cell killing, carcinogenesis, or mutation. There are many kinds of DSBs, varying in the distance between the breaks on the two DNA strands and the kinds of end groups formed. Their yield in irradiated cells is about 0.04 times that of SSBs, and they are induced linearly with dose, indicating that they are formed by single tracks of ionizing radiation.
in the next section, the interaction of two DSBs may result in cell killing, carcinogenesis, or mutation. There are many kinds of DSBs, varying in the distance between the breaks on the two DNA strands and the kinds of end groups formed. Their yield in irradiated cells is about 0.04 times that of SSBs, and they are induced linearly with dose, indicating that they are formed by single tracks of ionizing radiation.
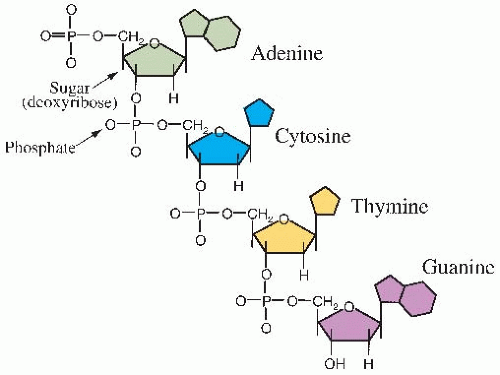 FIGURE 2.1 The structure of a single strand of DNA. |
Both free radicals and direct ionizations may be involved in the formation of the type of strand break illustrated in Figure 2.2D. As described in Chapter 1, the energy from ionizing radiations is not deposited uniformly in the absorbing medium but is located along the tracks of the charged particles set in motion—electrons in the case of x-or γ-rays, protons and α-particles in the case of neutrons. Radiation chemists speak in terms of “spurs,” “blobs,” and “short tracks.” There is, of course, a full spectrum of energy event sizes, and it is quite arbitrary to divide them into just three categories, but it turns out to be instructive. A spur contains up to 100 eV of energy and involves, on average, three ion pairs. In the case
of x- or γ-rays, 95% of the energy deposition events are spurs, which have a diameter of about 4 nm, which is about twice the diameter of the DNA double helix (Fig. 2.3). Blobs are much less frequent for x- or γ-rays; they have a diameter of about 7 nm and contain on average about 12 ion pairs with an energy range of 100-500 eV (Fig. 2.3). Because spurs and blobs have dimensions similar to the DNA double helix, multiple radical attacks occurs if they overlap the DNA helix. There is likely to be a wide variety of complex lesions, including base damage as well as DSBs. The term locally multiply damaged site was initially coined by John Ward to describe this phenomenon, but it has been replaced with the term clustered lesion. Given the size of a spur and the diffusion distance of hydroxyl free radicals, the clustered lesion could be spread out up to 20 base pairs. This is illustrated in Figure 2.3, in which a DSB is accompanied by base damage and the loss of genetic information.
of x- or γ-rays, 95% of the energy deposition events are spurs, which have a diameter of about 4 nm, which is about twice the diameter of the DNA double helix (Fig. 2.3). Blobs are much less frequent for x- or γ-rays; they have a diameter of about 7 nm and contain on average about 12 ion pairs with an energy range of 100-500 eV (Fig. 2.3). Because spurs and blobs have dimensions similar to the DNA double helix, multiple radical attacks occurs if they overlap the DNA helix. There is likely to be a wide variety of complex lesions, including base damage as well as DSBs. The term locally multiply damaged site was initially coined by John Ward to describe this phenomenon, but it has been replaced with the term clustered lesion. Given the size of a spur and the diffusion distance of hydroxyl free radicals, the clustered lesion could be spread out up to 20 base pairs. This is illustrated in Figure 2.3, in which a DSB is accompanied by base damage and the loss of genetic information.
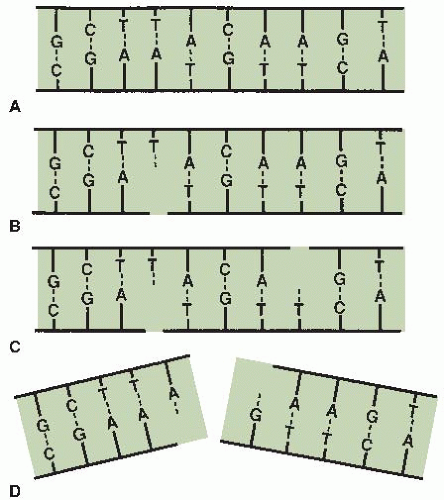 FIGURE 2.2 Diagrams of single- and double-strand DNA breaks caused by radiation. A: Two-dimensional representation of the normal DNA helix. The base pairs carrying the genetic code are complementary (i.e., adenine pairs with thymine, guanine pairs with cytosine). B: A break in one strand is of little significance because it is repaired readily using the opposite strand as a template. C: Breaks in both strands, if well separated, are repaired as independent breaks. D: If breaks occur in both strands and are directly opposite or separated by only a few base pairs, this may lead to a double-strand break in which the chromatin snaps into two pieces. (Courtesy of Dr. John Ward.) |
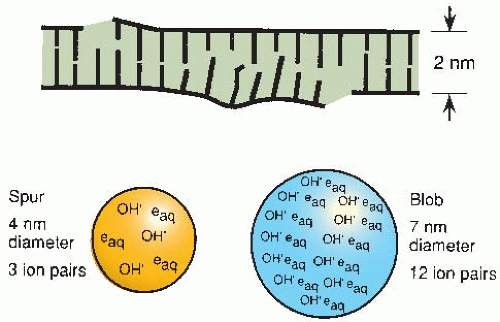 FIGURE 2.3 Illustration of a locally multiply damaged site. Energy from x-rays is not absorbed uniformly but tends to be localized along the tracks of charged particles. Radiation chemists speak in terms of spurs and blobs, which contain several ion pairs and have dimensions comparable to the DNA double helix. A double-strand break is likely to be accompanied by extensive base damage. John Ward coined the term locally multiply damaged site to describe this phenomenon. |
In the case of densely ionizing radiations, such as neutrons or α-particles, a greater proportion of blobs are produced. The damage produced, therefore, is qualitatively different from that produced by x- or γ-rays and it is much more difficult for the cell to repair.
▪ MEASURING DNA STRAND BREAKS
Over the years, various techniques have been used to measure DNA strand breaks, including sucrose gradient sedimentation, alkaline and neutral filter elution, nucleoid sedimentation, pulsed-field gel electrophoresis (PFGE), and single-cell gel electrophoresis (also known as the comet assay). Of these techniques, PFGE and single-cell gel electrophoresis are still used to measure DNA strand breaks. In addition to these past techniques, radiation-induced nuclear foci has become a popular approach to visualize DNA damage through the recruitment of DNA repair proteins to sites of DNA damage.
PFGE is the method most widely used to detect the induction and repair of DNA DSBs. It is based on the electrophoretic elution of DNA from agarose plugs within which irradiated cells have been embedded and lysed. PFGE allows separation of DNA fragments according to size in the megabase-pair range, with the assumption that DNA DSBs are induced randomly. The fraction of DNA released from the agarose plug is directly proportional to dose (Fig. 2.4A). The kinetics of DNA DSB rejoining exhibit a fast initial rate, which then decreases with repair time. The most widely accepted description of this kinetic behavior uses two first-order components (fast and slow) plus some fraction of residual DSBs. Studies have supported the finding that rejoining of incorrect DNA ends originates solely from slowly rejoining DSBs, and this subset of radiation-induced DSBs is what is manifested as chromosomal damage (i.e., chromosome translocations and exchanges).
Single-cell electrophoresis (comet assay) has the advantage of detecting differences in DNA damage and repair at the single-cell level. This is particularly advantageous for biopsy specimens from tumors in which a relatively small number of cells can be assayed to determine DNA damage and repair. Similar to PFGE (described earlier), cells are exposed to ionizing radiation, embedded in agarose, and lysed under neutral buffer conditions to quantify induction
and repair of DNA DSBs. To assess DNA SSBs and alkaline-sensitive sites, lysis is performed with an alkaline buffer. If the cells are undamaged, the DNA remains compact and does not migrate. If the cell has incurred DNA DSBs, the amount of damage is directly proportional to the migration of DNA in the agarose. As a result of the lysis and electrophoresis conditions, the fragmented DNA that migrates takes the appearance of a comet’s tail (Fig. 2.4B). This assay has high sensitivity and specificity for SSBs and alkaline sensitive sites and to a lesser degree DNA DSBs. By changing the lysis conditions from an alkaline to a neutral pH, the comet technique can be used to measure DNA DSB repair.
and repair of DNA DSBs. To assess DNA SSBs and alkaline-sensitive sites, lysis is performed with an alkaline buffer. If the cells are undamaged, the DNA remains compact and does not migrate. If the cell has incurred DNA DSBs, the amount of damage is directly proportional to the migration of DNA in the agarose. As a result of the lysis and electrophoresis conditions, the fragmented DNA that migrates takes the appearance of a comet’s tail (Fig. 2.4B). This assay has high sensitivity and specificity for SSBs and alkaline sensitive sites and to a lesser degree DNA DSBs. By changing the lysis conditions from an alkaline to a neutral pH, the comet technique can be used to measure DNA DSB repair.
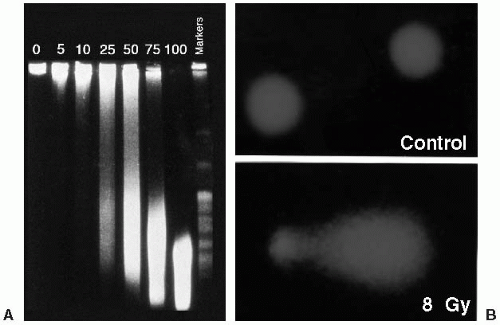 FIGURE 2.4 A: The effect of ionizing radiation on DNA strand break induction as measured by pulsed-field gel electrophoresis. As the dose of ionizing radiation increases from 5 to 100 Gy, the size of the DNA fragments as detected by ethidium bromide staining decreases. Thus, more DNA enters the gel with increasing dose of ionizing radiation. In these experiments, cells were embedded in agarose and irradiated on ice to eliminate the effects of repair. The number above each lane refers to the dose in Gy to which each group of cells was exposed. (Courtesy of Dr. Nicholas Denko.) B: Photomicrograph of control and 8-Gy irradiated cells as detected by the comet assay. Unirradiated cells possess a nearspherical appearance, whereas the fragmented DNA in irradiated cells gives the appearance of a comet when stained with ethidium bromide. (Courtesy of Drs. Ester Hammond and Mary Jo Dorie.) |
Both of these assays are cell based, where DNA in cells is much more resistant to damage by radiation than would be expected from studies on free DNA. There are two reasons for this: (1) the presence in cells of low-molecular-weight scavengers that mop up some of the free radicals produced by ionizing radiation, and (2) the physical protection afforded the DNA by packaging with proteins such as histones. Certain regions of DNA, particularly actively transcribing genes, appear to be more sensitive to radiation, and there is some evidence also of sequence-specific sensitivity.
DNA damage-induced nuclear foci (radiation-induced foci assay) in response to ionizing radiation represents complexes of signaling and repair proteins that localize to sites of DNA strand breaks in the nucleus of a cell. There are several advantages of assaying for foci formation over other techniques to measure DNA strand breaks, which include the ease of the protocol and that it can be carried out on both tissue sections and individual cell preparations. Technically, cells/tissues are incubated with a specific antibody raised to the signaling/repair protein of interest, and binding of the antibody is then detected with a secondary antibody, which also carries a fluorescent tag. Fluorescence microscopy detects the location and intensity of the tag, which can then be quantified.
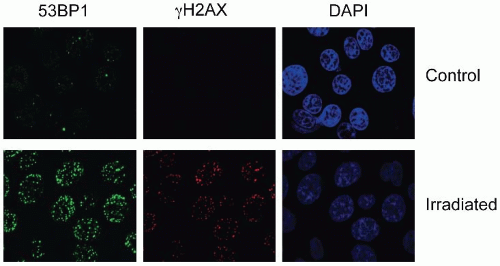 FIGURE 2.5 Photomicrograph of nuclear foci in control and 2-Gy irradiated cells as detected by staining with antibodies to 53BP1 (green) and γH2AX (red). Cells were also stained with the nuclear stain 4′,6-diamidino-2-phenylindole (DAPI) to show the location of nuclei. Without DNA strand breaks, there is little staining with γH2AX and 53BP1 in foci. In contrast, staining for both proteins increases significantly after 2 Gy. (Courtesy of Dr. Ester Hammond.) |
The most commonly assayed proteins for foci formation are γH2AX and 53BP1 (Fig. 2.5). H2AX is a histone protein, which is rapidly phosphorylated in response to damage to form γH2AX. Staining for the unmodified histone (H2AX) gives a pan nuclear stain or unchanging band on a western blot while γH2AX is rapidly induced on a western blot in response to stress and can be seen to form discreet nuclear foci in damaged cells (Fig. 2.5). 53BP1 also becomes phosphorylated in response to stress and forms nuclear foci at the sites of DNA DSBs. In this case, antibodies to either the phosphorylated or unmodified form can be used to detect DSBs as the protein relocalizes to the damaged chromatin (i.e., it is not already part of the chromatin as is the case for H2AX). DNA damage-induced increases in γH2AX or phosphorylated-53BP1 can also be quantified by flow cytometry. Other proteins also form foci in response to damage such as ataxia-telangiectasia mutated (ATM), replication protein A (RPA), RAD51, BRCA1 (discussed in subsequent sections).
Several γH2AX or 53BP1 foci that form in a damaged cell directly correlate with several DSBs present. If this value is measured over time, then it also reflects the kinetics of repair (i.e., as the DSBs are repaired, the number of foci decreases). Recently, BRCA1 and RAD51, two proteins involved in the repair of DNA damage by homologous recombination, have been used as biomarkers in a small pilot study by Willers et al. to detect repair defects in breast cancer biopsies.
▪ DNA REPAIR PATHWAYS
Mammalian cells have developed specialized pathways to sense, respond to, and repair base damage, SSBs, DSBs, sugar damage, and DNA-DNA crosslinks. Research from yeast to mammalian cells has demonstrated that the mechanisms used to repair ionizing radiation-induced base damage are different from the mechanisms used to repair DNA DSBs. In addition, different repair pathways are used to repair DNA damage, depending on the stage of the cell cycle.
Much of our knowledge of DNA repair is the result of studying how mutations in individual genes result in radiation hypersensitivity. Radiation-sensitive mutants identified from yeast and mammalian cells appear either to be directly involved in the repair process or to function as molecular checkpoint-controlling elements. The pathways involved in the repair of base damage, SSBs, DSBs, sugar damage, and DNA-DNA crosslinks are discussed in the next sections and represent a simplified representation of our current state of understanding.
In Chapter 18, the syndromes associated with mutations in genes involved in sensing DNA damage or repairing DNA damage are discussed in more detail.
In Chapter 18, the syndromes associated with mutations in genes involved in sensing DNA damage or repairing DNA damage are discussed in more detail.
Base Excision Repair
Base damage is repaired through the base excision repair (BER) pathway illustrated in Figure 2.6. Bases on opposite strands of DNA must be complementary; adenine (A) pairs with thymine (T), and guanine (G) pairs with cytosine (C). U therefore represents a putative single-base mutation that is first removed by a glycosylase/DNA lyase (Fig. 2.6A). Removal of the base is followed by the removal of the sugar residue by an apurinic endonuclease 1 (APE1), then replacement with the correct nucleotide by DNA polymerase β, and completed by DNA ligase III-XRCC1– mediated ligation. If more than one nucleotide is to be replaced (illustrated by the putative mutation UU in Fig. 2.6B), then the complex of replication factor C (RFC)/proliferating cell nuclear antigen (PCNA)/DNA polymerase δ/ε performs the repair synthesis, the overhanging flap structure is removed by the flap endonuclease 1 (FEN1), and DNA strands are sealed by ligase I (Fig. 2.6B). Although ionizing radiation-induced base damage is efficiently repaired, defects in BER may lead to an increased mutation rate, but usually do not result in cellular radiosensitivity. One exception to this is the mutation of the x-ray cross complementing factor 1 (XRCC1) gene, which confers about a 1.7-fold increase in radiation sensitivity. However, the radiation sensitivity of XRCC1-deficient cells may come from XRCC1‘s potential involvement in other repair processes such as SSBs.
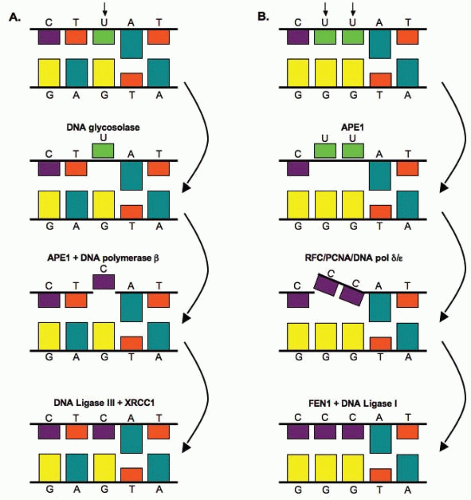 FIGURE 2.6 Base excision repair pathways. A: Base excision repair of a single nucleotide. Bases on opposite strands must be complementary; adenine (A) pairs with thymine (T), and guanine (G) pairs with cytosine (C). U represents a putative mutation that is first removed through a DNA glycosylase-mediated incision step. B: Base excision repair of multiple nucleotides. In this case, the double UU represents a putative mutation that is first removed through apurinic endonuclease 1 (APE1). See text for details. |
Nucleotide Excision Repair
Nucleotide excision repair (NER) removes bulky adducts in the DNA such as pyrimidine dimers. The process of NER can be subdivided into two pathways: global genome repair (GGR or GG-NER) and transcription-coupled repair (TCR or TC-NER). The process of GG-NER is genome-wide (i.e., lesions can be removed from DNA that encodes or does not encode for genes). In contrast, TC-NER only removes lesions in the DNA strands of actively
transcribed genes. When a DNA strand that is being actively transcribed becomes damaged, the RNA polymerase can block access to the site of damage and hence prevents DNA repair. TC-NER has evolved to prevent this blockade by RNA polymerase by effectively removing it from the site of damage to allow the repair proteins access. The mechanism of GG-NER and TC-NER differs only in the detection of the lesion; the remainder of the pathway used to repair the damage is the same for both. The essential steps in this pathway are (1) damage recognition; (2) DNA incisions that bracket the lesion, usually between 24 and 32 nucleotides in length; (3) removal of the region containing the adducts; (4) repair synthesis to fill in the gap region; and (5) DNA ligation (Fig. 2.7). Mutation in NER genes does not lead to ionizing radiation sensitivity. However, defective NER increases sensitivity to UV-induced DNA damage and anticancer agents such as alkylating agents that induce bulky adducts. Germline mutations in NER genes lead to human DNA repair deficiency disorders such as xeroderma pigmentosum in which patients are hypersensitive to ultraviolet light.
transcribed genes. When a DNA strand that is being actively transcribed becomes damaged, the RNA polymerase can block access to the site of damage and hence prevents DNA repair. TC-NER has evolved to prevent this blockade by RNA polymerase by effectively removing it from the site of damage to allow the repair proteins access. The mechanism of GG-NER and TC-NER differs only in the detection of the lesion; the remainder of the pathway used to repair the damage is the same for both. The essential steps in this pathway are (1) damage recognition; (2) DNA incisions that bracket the lesion, usually between 24 and 32 nucleotides in length; (3) removal of the region containing the adducts; (4) repair synthesis to fill in the gap region; and (5) DNA ligation (Fig. 2.7). Mutation in NER genes does not lead to ionizing radiation sensitivity. However, defective NER increases sensitivity to UV-induced DNA damage and anticancer agents such as alkylating agents that induce bulky adducts. Germline mutations in NER genes lead to human DNA repair deficiency disorders such as xeroderma pigmentosum in which patients are hypersensitive to ultraviolet light.
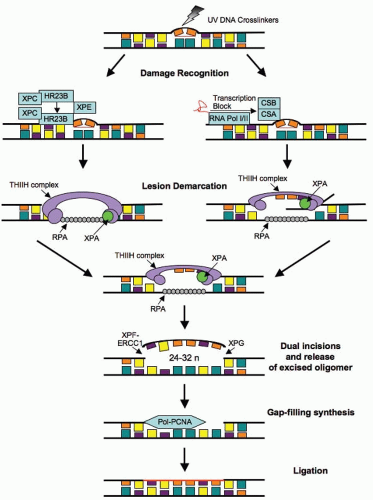 FIGURE 2.7 Nucleotide excision repair pathways. The two subpathways of NER, GG-NER/GGR (global genome repair) and TC-NER/TCR (transcription-coupled repair), differ at the initial damage recognition step. GGR uses the XPC-XPE protein complexes, whereas in TCR, the NER proteins are recruited by the stalled RNA polymerase in cooperation with CSB and CSA. Following recognition, the lesion is demarked by binding of the transcription factor IIH (TFIIH) complex, XPA and RPA. The TFIIH complex helicase function unwinds the DNA and generates an open stretch around the lesion, at which point the XPG and XPF-ERCC1 endonucleases make incisions at the 3′ and 5′ ends, respectively, releasing a 24-32 oligomer. The resulting gap is filled by the polymerases δ/ε aided by RFC and PCNA and the strand is finally ligated. (XPC, xeroderma pigmentosum, complementation group C; XPE, xeroderma pigmentosum, complementation group E; CSB, cockayne Syndrome B gene; CSA, cockayne Syndrome A gene; XPG, xeroderma pigmentosum, complementation group G; XPF, xeroderma pigmentosum, complementation group F; ERCC1, excision repair cross-complementation group 1 gene; RFC, replication factor C; PCNA, proliferating cell nuclear antigen.) |
DNA Double-Strand Break Repair
In eukaryotic cells, DNA DSBs can be repaired by two basic processes: homologous recombination repair (HRR), which requires an undamaged DNA strand as a participant in the repair as a template, and nonhomologous end-joining (NHEJ), which mediates end-to-end joining. In lower eukaryotes such as yeast, HRR is the predominant pathway used for repairing DNA DSBs. Homologous recombination is an error-free process because
repair is performed by copying information from the undamaged homologous chromatid/chromosome. In mammalian cells, the choice of repair is biased by the phase of the cell cycle and by the abundance of repetitive DNA. HRR occurs primarily in the late S/G2 phase of the cell cycle, when an undamaged sister chromatid is available to act as a template, whereas NHEJ occurs in the G1 phase of the cell cycle, when no such template exists (Fig. 2.8). NHEJ is error prone and probably accounts for many of the premutagenic lesions induced in the DNA of human cells by ionizing radiation. However, it is important to keep in mind that NHEJ and HRR are not mutually exclusive, and both have been found to be active in the late S/G2 phase of the cell cycle, indicating that other as-yet-unidentified factors, in addition to cell cycle phase, are important in determining what repair program is used.
repair is performed by copying information from the undamaged homologous chromatid/chromosome. In mammalian cells, the choice of repair is biased by the phase of the cell cycle and by the abundance of repetitive DNA. HRR occurs primarily in the late S/G2 phase of the cell cycle, when an undamaged sister chromatid is available to act as a template, whereas NHEJ occurs in the G1 phase of the cell cycle, when no such template exists (Fig. 2.8). NHEJ is error prone and probably accounts for many of the premutagenic lesions induced in the DNA of human cells by ionizing radiation. However, it is important to keep in mind that NHEJ and HRR are not mutually exclusive, and both have been found to be active in the late S/G2 phase of the cell cycle, indicating that other as-yet-unidentified factors, in addition to cell cycle phase, are important in determining what repair program is used.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


