Fig. 25.1
Parkinson disease. 72-year-old woman. (a) T2-weighted image, axial; (b) T1-weighted image, sagittal; (c) T2-weighted image, coronal; (f, g) Neuromelanin images (f: upper pons, g: midbrain); and (d, e) neuromelanin image of normal adult (f: upper pons, g: midbrain). T1-weighted image and T2-weighted images show no characteristic abnormal signals in the basal ganglia or other structures (a–c). Midsagittal image does not show any characteristic pattern of atrophy (b). On coronal image, cerebellar hemisphere, superior cerebellar peduncle, and middle cerebellar peduncle do not show any atrophy (c). On neuromelanin images, comparisons of Parkinson patients to normal adults show reduced hyperintensity in locus coeruleus (d) and substantia nigra pars compacta (e), suggesting depletion of neuromelanin-containing neurons. Compare with those in a normal adult (f, g)
25.2.2 Multiple System Atrophy with Predominant Parkinsonism (MSA-P: Striatonigral Degeneration)
Multiple system atrophy (MSA) is a sporadic neurodegenerative disorder that includes different disease entities showing degeneration within the pyramidal tract, extrapyramidal system, cerebellum, and autonomic system. The histopathological characteristics are the formation of alpha-synuclein-positive glial cytoplasmic inclusions in oligodendroglial cells. Details of this disorder will be summarized in a later section.
Striatonigral degeneration (MSA with predominant Parkinsonism: MSA-P) is a part of MSA and the major feature of the disease is Parkinsonism, for which anti-Parkinson agents are not effective. Degeneration of the extrapyramidal system, especially striatum and substantia nigra, is the hallmark of this disease. Degeneration is prominent in the posterolateral region of the putamen, in which loss of neurons with replacement by glia cells and iron deposition can be observed.
The most characteristic MR findings for MSA-P are changes in the putamen. The putamen shows atrophic changes and abnormal signal. The lateral borders of the putamen become angulated in contrast to the curved border of the normal putamen (Fig. 25.2).
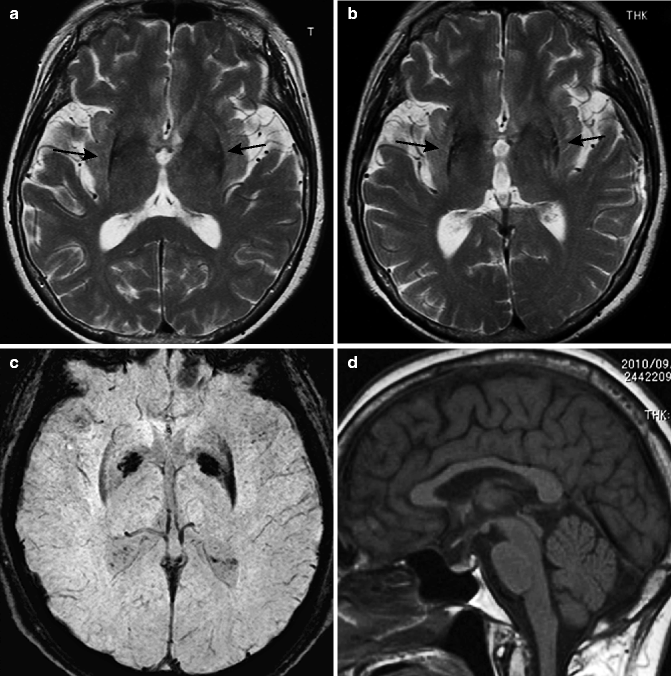
Fig. 25.2
MSA with predominant parkinsonism (MSA-P). 57-year-old man with dysphagia and dyspnea. (a) T2-weighted image, axial (2 years prior); (b) T2-weighted image, axial; (c) susceptibility-weighted image, axial; (d) T1-weighted image, sagittal; (e, f) T2-weighted images, coronal. Putaminal atrophy and hyperintensity on T2-weighted images in dorsolateral aspect of putamen can be seen especially on dorsolateral aspect (a, b). Lower signal intensity can be seen medial to hyperintensity. Lateral margins of putamen become linear on axial and coronal image (arrows) (a, b, e). Hypointensity is prominent on susceptibility-weighted image (c). Note the progression of putaminal atrophy in 2 years (a, b). Brainstem atrophy is not prominent in this case (d). Volume of the cerebral hemisphere and middle cerebellar peduncle is also preserved in this case (f)
25.2.3 Progressive Supranuclear Palsy
Progressive supranuclear palsy (PSP) is a tauopathy in which there is deposition of tau protein within neurons or glial cells. Tau protein is a microtubule-associated protein that interacts with tubulin to promote the building of microtubules from tubulin. Tau protein also acts as a stabilizer for microtubules. Abnormal phosphorylation of tau protein, however, results in the self-assembly of tangles and deposition of these aggregated filaments. There are two isoforms of tau protein, a 3-repeat tau and a 4-repeat tau, according to the number of microtubule-binding domains. Both PSP and corticobasal degeneration (CBD, see below) involve accumulation of the abnormal 4-repeat tau isoform in neurons and glial cells. Although CBD and PSP have different clinical features, they have overlapping clinical, pathological, genetic, and biochemical features, and the differential diagnosis is often complicated, even at autopsy. Thus, these two disorders may represent two points on the same disease spectrum. The pathological changes of PSP are distributed in the basal ganglia and brainstem, especially in the subthalamic nucleus, superior colliculus of the midbrain, pars compacta of the substantia nigra, red nucleus, putamen, and dentate nucleus.
Symptoms of PSP tend to appear in the sixth or seventh decade of life, with gait disturbance being the first symptom in most cases. The characteristic symptom of this disease is supranuclear ocular motility disorder with spared oculocephalic reflex. Vertical ocular motion is impaired, particularly in the downward gaze. As the disease progresses, horizontal gaze is also impaired, and the oculocephalic reflex is lost at the end stage of the disease. Symptoms of PSP also include Parkinsonism, such as bradykinesia or rigidity, for which anti-Parkinson agents are not effective.
The most characteristic imaging finding for PSP is atrophy in the superior colliculus and tectum of the midbrain, which results in the dilation of the cerebral aqueduct and the posterior part of the third ventricle (Yagishita and Oda 1996). The volume of the basal part of the pons is preserved. Hyperintensity around the aqueduct on T2-weighted images has also been reported (Aiba et al. 1997). Several methods for quantitative evaluation of brainstem atrophy have been reported. In one report that compared 50 patients with various Parkinsonian syndromes (Parkinson disease, PSP, and MSA-P) with control subjects, patients with PSP had significantly lower anteroposterior diameters of the suprapontine midbrain (mean, 13.4 mm) than patients with Parkinson disease (mean, 18.5 mm) and control subjects (mean, 18.2 mm). However, midbrain diameters of patients with MSA-P were also significantly lower than those of control subjects and patients with Parkinson disease, with individual values showing overlap between the PSP, Parkinson disease, and control groups (Warmuth-Metz et al. 2001). Oba et al. reported that the area of the midbrain and pons on midsagittal images can differentiate PSP from other degenerative diseases and normal aging (Oba et al. 2005). Their measurements showed that the midbrain area in PSP patients was significantly smaller than that in patients with Parkinson disease, MSA-P, and age-matched controls (Fig. 25.3).
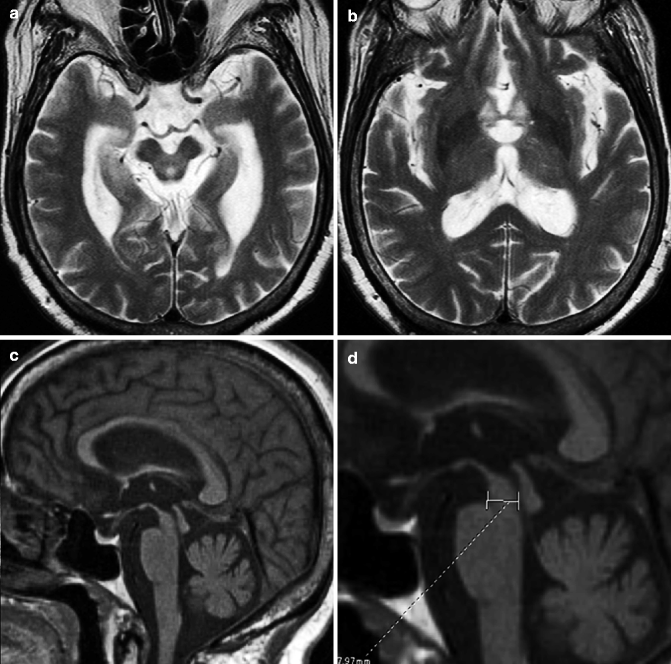
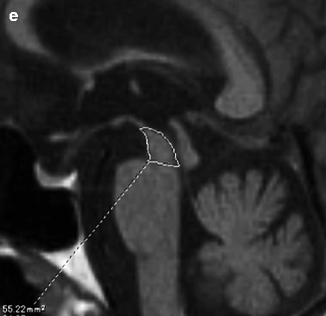
Fig. 25.3
Progressive supranuclear palsy. 67-year-old woman with gait disturbance. (a, b) T2-weighted images, axial; (c–e) T1-weighted images, sagittal. Severe atrophy in superior colliculus and tectum of midbrain can be seen on both axial (a) and sagittal image (c). Volume of putamen (b) and basal part of pons is preserved (c). Anteroposterior diameter of the suprapontine midbrain was reduced to 7.9 mm (d). The area of the midbrain and pons on midsagittal images was 55 mm2 which was significantly smaller than in age-matched controls (e)
25.2.4 Corticobasal Degeneration
Corticobasal degeneration (CBD) is a tauopathy and a progressive neurodegenerative disease with both motor and cognitive dysfunction. Similar to PSP, CBD also shows accumulation of the abnormal 4-repeat tau isoform in neurons and glial cells. The pathological changes of CBD are characterized by asymmetric neuronal loss in the frontal and parietal lobes, in which ballooned neurons can be seen. Degeneration of the basal ganglia is prominent in the globus pallidus, and the striatum, thalamus, and substantia nigra also show degeneration.
The symptoms of CBD usually appear in the sixth to eighth decade of life with asymmetrical extrapyramidal symptoms and cortical symptoms including apraxia, cortical sensory deficits, and alien limb phenomenon.
Imaging findings in CBD patients reveal the primary characteristic changes associated with CBD (Tokumaru et al. 2009). There is asymmetric paracentral cortical and basal ganglia atrophy (Fig. 25.4). Curvilinear T2 prolongation in the posterolateral putamen on the side contralateral to the symptoms can also be seen (Tokumaru et al. 1996). Midbrain tegmentum atrophy is severe, and degeneration in this area is correlated with disease severity, similar to findings in patients with PSP. T1-weighted image shows symmetric high-intensity signals in the subthalamic nucleus, which is also similar to findings in patients with PSP. Diffuse high-intensity signals in the subcortical white matter on T2-weighted images and fluid-attenuated inversion recovery (FLAIR) can also be seen (Koyama et al. 2007). Corresponding to the sites of these white matter lesions, myelin sheath staining is reportedly decreased, and these sites stain positively for antiphosphorylated tau antibody.
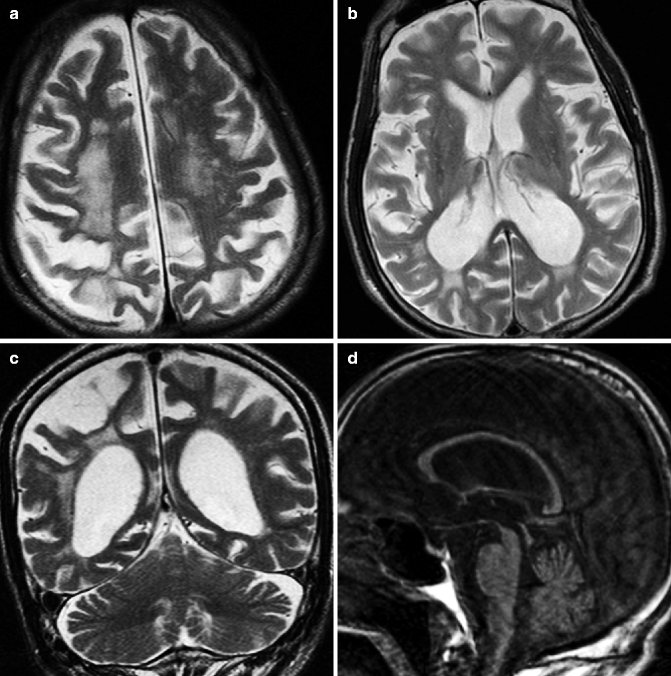
Fig. 25.4
Corticobasal degeneration. 72 year-old man with gait disturbance and rigidity on left side. (a, b) T2-weighted images, axial; (c) T2-weighted image, coronal; (d) T1-weighted image, sagittal. Right dominant asymmetric paracentral cortical atrophy can be seen (a, c). Diffuse high-intensity signals in subcortical white matter on T2-weighted images can also be seen (a). There is also basal ganglia atrophy and curvilinear T2 prolongation in posterolateral putamen (b). Slight midbrain tegmentum atrophy is noted on midsagittal sections (d)
25.3 Disorders with Involuntary Movement
25.3.1 Huntington Disease
Huntington disease is a progressive degenerative disease that results from an autosomal dominant mutation that typically becomes apparent in the fourth to sixth decade of life. Huntington is a triplet repeat disease in the Huntington gene, with CAG repeat expansion on the short arm of chromosome 4p16.3. Patients with Huntington disease exhibit involuntary movements. Symptoms start with chorea-like involuntary movements and progress to psychiatric symptoms, character change, and eventually dementia. The characteristic pathological change of Huntington disease is neuronal loss and gliosis in the striatum, especially in the caudate nucleus. The cerebral cortex also shows neuronal loss in the pyramidal cell layers.
Images from patients with Huntington disease show dilation of the anterior horns of the lateral ventricles due to volume loss in the caudate nucleus (Fig. 25.5). The putamen also shows atrophic changes. Significant striatal atrophy can be seen many years prior to the clinical onset of the disease (Aylward et al. 2004). Occasionally, hyperintensity is visible in the striatum on T2-weighted images. The hyperintensity is detectable because of gliosis. Low signal can also occur in the striatum due to iron deposition.
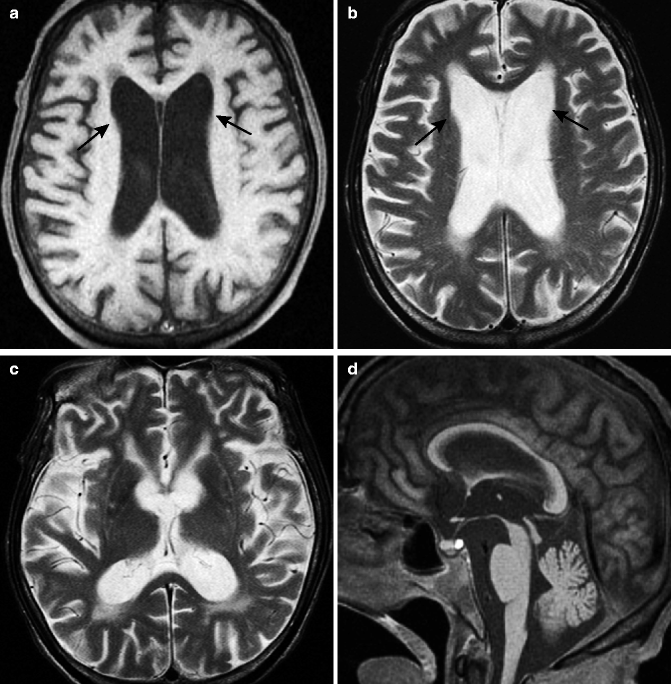
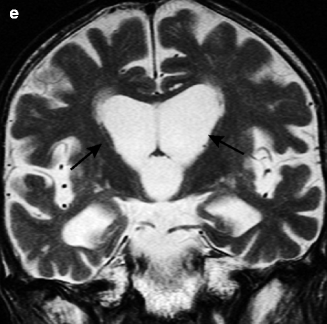
Fig. 25.5
Huntington disease. 68-year-old man with involuntary movement. (a) T1-weighted image, axial; (b, c) T2-weighted images, axial; (c) T1-weighted image, sagittal coronal; (d) T2-weighted image, coronal. Anterior horns of lateral ventricle are dilated and volume loss of caudate nucleus can be seen (arrows) (a, b, e). Atrophic changes and hyperintensity in putamen can be seen on T2-weighted image (c)
25.3.2 Chorea Acanthocytosis
Chorea acanthocytosis is one type of neuroacanthocytosis that shows both abnormal red blood cells called acanthocytes and neuronal symptoms such as seizures and movement dysfunction. A mutation in the VPS13A gene on chromosome 9q21 is known to cause this disorder. This group of diseases consists of chorea acanthocytosis as well as McLeod syndrome and PKAN (pantothenate kinase-associated neurodegeneration). Chorea acanthocytosis is an autosomal recessive disorder, and onset is generally in the second to sixth decade of life. The clinical symptoms include chorea, behavioral changes, epilepsy, muscle degeneration, and increased serum creatine kinase levels.
The major imaging findings associated with this disorder are atrophy of the caudate nucleus with dilation of the anterior horns of the lateral ventricles, which is milder compared to that observed in cases of Huntington disease. Atrophy is also seen in the putamen. MRI also commonly shows hyperintensity in the caudate and putamen on T2-weighted images (Gradstein et al. 2005). Hippocampal sclerosis and atrophy are also apparent (Al-Asmi et al. 2005).
25.3.3 Gilles de la Tourette Syndrome
Gilles de la Tourette syndrome is a disorder that typically begins in childhood with multiple motor tics that are repetitive, stereotyped, involuntary movements, usually occurring with one or more vocal tics. The definitive pathophysiology is not well known; however, since antidopaminergic agents are effective for suppressing the symptoms, hyperactivity of the dopaminergic system is suspected to play a role.
Several reports about MRI morphologic studies have been published. Regional abnormalities in gray matter, especially in the basal ganglia region, prefrontal to the parieto-occiptal cortex, and in the cerebellum, have been reported (Peterson et al. 2003; Peterson 2001; Sowell et al. 2008; Tobe et al. 2010). High-frequency stimulation by deep electrodes in the centromedian nucleus-substantia periventricularis-nucleus ventro-oralis internus cross point is reported to reduce tic severity (Ackermans et al. 2011) (Fig. 25.6).
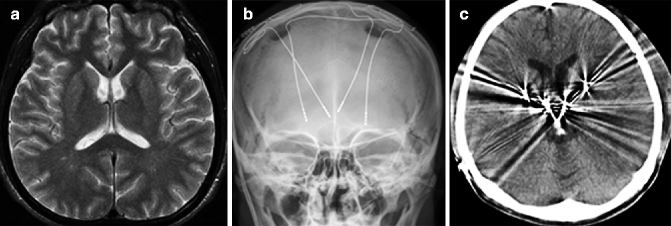
Fig. 25.6
Gilles de la Tourette syndrome. 35-year-old man with tic movements and mental retardation. (a) T2-weighted image, axial; (b) skill X ray; (c) CT after placement of deep electrodes. Slight abnormal signal in basal ganglia can be seen on T2-weighted image, although, signal changes in the cortex are not obvious. High-frequency stimulation of putamen and thalamus using deep, implanted electrodes has reduced tic severity
25.3.4 Fahr Disease
Fahr disease (or idiopathic calcification of the basal ganglia) is a degenerative disease in which symmetric abnormal calcium deposition occurs in the basal ganglia, dentate nucleus, and cerebral white matter (Fig. 25.7). Histologically, the calcium deposition in Fahr disease is seen in the walls of capillaries, arterioles, venules, or in perivascular spaces and may be accompanied by adjacent neural degeneration and/or gliosis. There are a wide variety of clinical symptoms, including Parkinsonism, choreoathetotic-type movement disturbances, cognitive disorders, and cerebellar ataxia. There are two patterns in the onset of Fahr disease, including early onset (around 30 years old), in which the occurrence of movement disorders is minimal, and late onset (around 50 years old), which presents with dementia and movement dysfunction (Cummings et al. 1983). Differential diagnoses of Fahr disease using imaging methods include hypoparathyroidism or pseudohypoparathyroidism. Thus, measurements of serum calcium, phosphorus, and parathyroid hormone levels are useful for diagnosis (Avrahami et al. 1994).
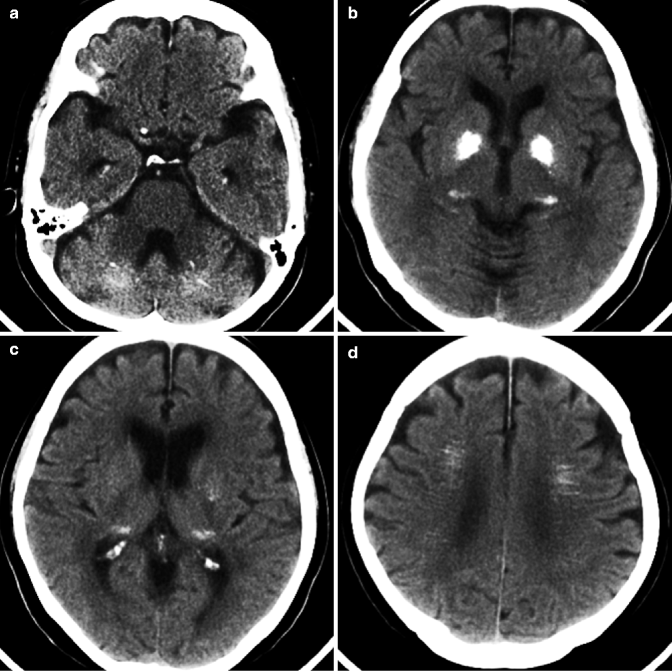
Fig. 25.7
Fahr disease. 78-year-old woman with dementia. (a–d) Axial view of plain CT. Abnormal calcium deposition can be seen in basal ganglia (b, c), dentate nucleus (a), and cerebral white matter (d)
25.3.5 Non-ketotic Hyperglycemic Chorea Hemiballismus
Non-ketotic hyperglycemic chorea-hemiballismus, diabetic hemichorea/hemiballism or hyperglycemic choreoathetosis is a rare complication of non-ketotic hyperglycemia in type 2 diabetes. Chorea and ballismus can be caused by various conditions in which the striatum, external segment of the globus pallidus, thalamus, or subthalamic nucleus is impaired, and dysfunction of inhibitory fibers to the thalamus via globus pallidus results in disinhibition of thalamic neurons. The underlying causes include neurodegenerative diseases, cerebrovascular disorders, infections, drugs, metabolic abnormalities, and tumors, as well as non-ketotic hyperglycemia.
MRI from patients with this disorder is characterized by hyperintensity of the striatum and/or globus pallidus on the side contralateral to the symptoms on T1-weighted images (Fig. 25.8). T2-weighted images usually do not show any signal abnormalities. Occasionally there are cases with the signal abnormality on T1-weighted images on both sides. In these cases, their symptoms are also bilateral. The signal abnormality is reversible, and T1 hyperintense lesions in the basal ganglia regress in the cases receiving follow-up MRI during the remission phase (Chang et al. 2010). There are some hypotheses for hyperintensity on T1-weighted images including reversible ischemic insult potentiated by hyperglycemia, petechial hemorrhage, myelin destruction, deposition of manganese, or microgliosis (Chang et al. 2010; Lai et al. 1996; Shan et al. 2007). However, no single definite mechanism has been proposed.
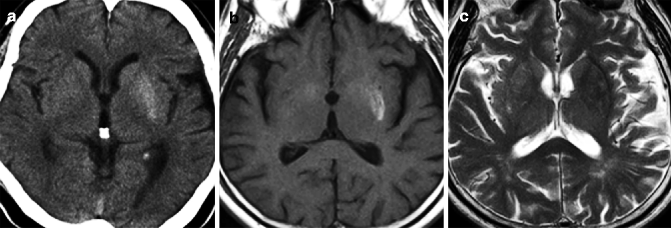
Fig. 25.8
Non-ketotic hyperglycemic chorea-hemiballismus. 67-year-old man with unilateral involuntary movement on right side and diabetes mellitus (blood sugar, 737 mg/day; HbA1c, 14.1 %). (a) CT, axial; (b) T1-weighted image, axial; (c) T2-weighted image, axial. Putamen on left side shows hyperintensity on T1-weighted image especially in posterior portion (b). CT also shows slight high density in left putamen (a). T2-weighted image does not show significant signal abnormality in posterior putamen (c)
25.4 Disorders Mainly Presenting Ataxia
Spinocerebellar degeneration (SCD) is a general term for the neurodegenerative disease in which cerebellar or spinal ataxia is the major symptom and systemic degeneration of the nucleus or tract within the cerebellum or spine takes place. SCD is divided into hereditary SCD and sporadic SCD. Hereditary SCD includes dentatorubral-pallidoluysian atrophy (DRPLA) and spinocerebellar ataxia (SCA). Sporadic SCD is further divided into multiple system atrophy (MSA), cortical cerebellar atrophy, and other syndromes with cerebellar atrophy.
25.4.1 Multiple System Atrophy
Multiple system atrophy is a sporadic, nonhereditary neurodegenerative disorder with cerebellar atrophy, autonomic disorder, and/or Parkinsonism in its course. MSA consists of olivopontocerebellar atrophy, which mainly presents cerebellar atrophy; striatonigral degeneration, which mainly presents Parkinsonism; and Shy-Drager syndrome, which mainly presents autonomic disorder. These three disorders are originally regarded as other entities; however, since argentaffin inclusion bodies, often called “glial cytoplasmic inclusions” in oligodendroglial cells, can be seen in these three disorders, these are regarded as a single entity. MSA predominantly presenting Parkinsonism is called MSA-P, MSA predominantly presenting cerebellar ataxia is called MSA-C, and the terminologies of olivopontocerebellar atrophy, striatonigral degeneration, and Shy-Drager syndrome are no longer used. The diagnostic criteria by the second consensus statement on MSA states that MSA is defined by neuropathologic findings of widespread and abundant central nervous system glial cytoplasmic inclusions that are positive for alpha-synuclein, in association with neurodegeneration in striatonigral or olivopontocerebellar structures (Gilman et al. 2008). Alpha-synuclein is one of the major components of intracellular fibrillary aggregates in the brains of a subset of neurodegenerative disorders, including Parkinson disease, dementia with Lewy bodies, multiple system atrophy, and Hallervorden-Spatz disease, which are referred to as alpha-synucleinopathies (Hasegawa et al. 2002). MSAs, including MSA-C and MSA-P, are categorized within a subgroup of these neurodegenerative disorders termed synucleinopathies, in which alpha-synuclein is polymerized into fibrils and accumulates in pathologic inclusions, such as Lewy bodies and glial cytoplasmic inclusions mentioned above. Although MSA is subdivided into two categories (MSA-P and MSA-C) according to the most prominent initial symptom, both symptoms, including Parkinsonism and cerebellar ataxia, become apparent with disease progress. Similarly, the pathological findings in the pons, which characterize MSA-C, and the findings in putamen, which characterize MSA-P, are both commonly seen in the terminal stage of disease.
25.4.1.1 MSA with Parkinsonism (MSA-P: Striatonigral Degeneration)
MSA-P and MSA-C share pathological characteristics when the disease progresses. Pathological findings in the putamen include atrophy of the putamen with loss the normal concavity at the lateral border of the putamen, degeneration with neuronal loss, gliosis, and iron accumulation. There are also findings in the pons, including degeneration within the pontine nucleus and transverse pontine fibers. The white matter of the cerebellar hemisphere, middle cerebellar peduncle, and olive also exhibit degeneration. Although the degree of degeneration varies considerably among cases, the degeneration in the putamen predominantly affects the posterior and lateral regions, with the more caudal regions left relatively intact in MSA-P.
On MRI, atrophy in the putamen, middle cerebellar peduncle, pons, or cerebellum are listed as findings that may indicate MSA-P in the diagnostic criteria by the second consensus statement on MSA (Gilman et al. 2008). The putaminal atrophy and the hyperintensity on T2-weighted images in the dorsolateral aspect of the putamen are more specific to MSA-P. The posterolateral region of the putamen, medial to the hyperintensity, shows lower signal intensity on T2-weighted images or T2*-weighted images, reflecting iron deposition (Fig. 25.2). Putaminal hyperintensity on T1-weighted images is also frequently observed in patients with MSA-P (Ito et al. 2009). The lateral margin of the putamen shows curvature in normal cases; however, the putamen in patients with MSA-P type atrophy shows a more linear margin. The putaminal abnormality starts in the dorsolateral aspect, and as the disease progresses, the signal change extends into the anterior part of the putamen. These putaminal findings may be unilateral in the early stages of MSA-P, and the symptoms of Parkinsonism may also be unilateral. In such a case, Parkinsonism can be seen on the side of the body contralateral to the putaminal change. As mentioned in the diagnostic criteria by the second consensus statement on MSA, findings within the pons and middle cerebellar peduncle can also be seen in MSA-P. Degeneration of transverse pontine fibers may present without degeneration of the putamen.
MSA with Cerebellar Ataxia (MSA-C: Olivopontocerebellar Degeneration)
The major pathological change in MSA-C is in the cerebellar afferent system, which includes the pontocerebellar tract and also the inferior olivary nucleus. There is prominent degeneration and atrophy in the basilar part of the pons due to degeneration in the pontine nuclei. The white matter in the cerebellar hemisphere is also affected, and Purkinje cells are lost in the cerebellar cortex. Degeneration of the inferior olivary nucleus is also apparent. In advanced cases, there are also changes within the striatonigral system, which includes the substantia nigra pars compacta and pars reticulata, which result in the autonomic symptoms, sharing similar pathological changes with MSA-P.
Images from patients with MSA-C show characteristic degeneration of the transverse pontine fibers and atrophy of the middle cerebellar peduncle. The degeneration of the transverse pontine fibers, which show high signal on T2-weighted images, appears in the earlier stages of the disease. The degenerative changes tend to appear in the midline portion of the basilar part of the pons and are followed by changes in the anterior portion of the medial lemniscus and middle cerebellar peduncle. In contrast, the signal in the pontine tegmentum is preserved as well as corticospinal tract, superior cerebellar peduncle, and dentate nucleus. As the disease progress and the degeneration of the transverse pontine fiber become prominent, the hyperintensity in the basilar part of the pons becomes crossed shape (cross bun sign) (Fig. 25.9). In addition to MSA-P, atrophy on MRI of the putamen, middle cerebellar peduncle, pons, or cerebellum is listed as image findings for the possible MSA-C in the diagnostic criteria by the second consensus statement on MSA (Gilman et al. 2008). White matter of cerebellar hemisphere may show hyperintensity on T2-weighted imaging.
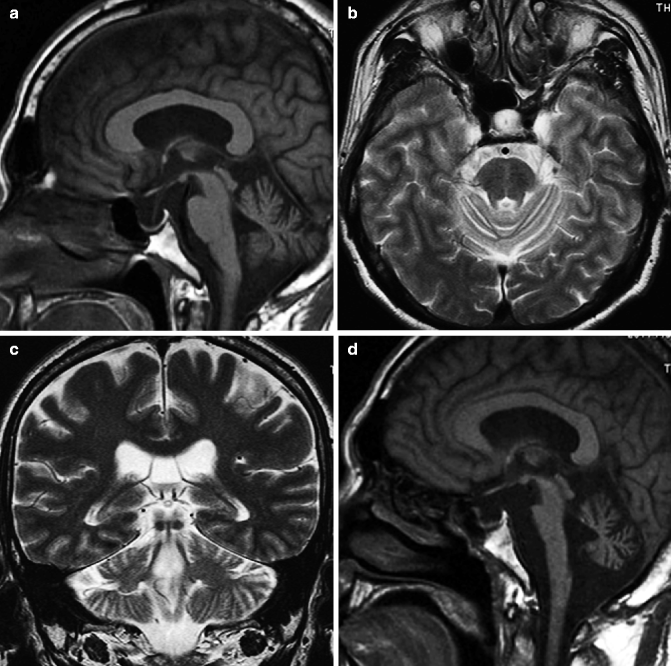
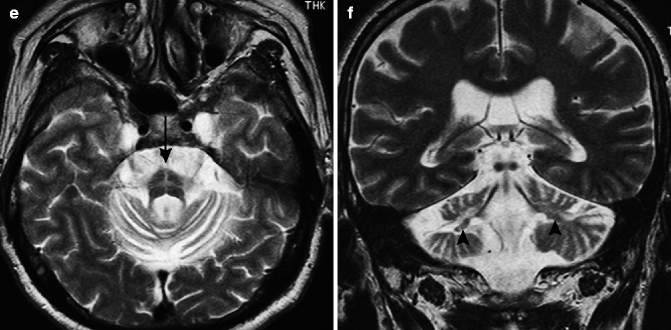
Fig. 25.9
MSA with cerebellar ataxia (MSA-C). 51-year-old man with gait disturbance. (a) T1-weighted image, sagittal; (b) T2-weighted image, axial; (c) T2-weighted image, coronal (a–c: 4 years prior to d–f); (d) T1-weighted image, sagittal; (e) T2-weighted image, axial; (f) T2-weighted image, coronal. Midsagittal image shows volume loss of the ventral portion of the pons (a, d). Degeneration of transverse pontine fibers shows high signal on T2-weighted image at midline portion in basilar part of pons appears in images from 4 years earlier (b). As disease progresses, hyperintensity in the basilar part of the pons assumes a cross shape (arrow) (cross bun sign, e). Atrophy of middle cerebellar peduncle is also prominent on follow-up images (arrowheads) (f). White matter of cerebellar hemispheres shows hyperintensity on T2-weighted image on follow-up (f)
25.4.1.2 Cortical Cerebellar Atrophy
Cortical cerebellar atrophy is a sporadic SCD and has also been called late cortical cerebellar atrophy due to the late onset of the symptoms. This disorder is also known as idiopathic cerebellar ataxia or sporadic adult-onset ataxia of unknown etiology. The pathological change associated with this disorder is limited to the cerebellar cortex and neuronal loss of Purkinje cells and cells in the inferior olivary nucleus (Ota et al. 2008). The major symptom of this disease is ataxia, which starts with an ataxic gait and ataxia in the lower extremities followed by ataxia in the upper extremities. There is no Parkinsonism and no autonomic symptoms. The typical onset age of this disorder is in the sixth to seventh decades of life, and it occurs predominantly in men.
The major imaging finding for this disorder is cerebellar atrophy in the gray matter (Fig. 25.10). The atrophy of the cerebellum is noticeable in the vermis, the anterior lobe, and the upper part of the posterior lobe. There is no sign of atrophy of the transverse pontine fibers or atrophy of the brainstem. A loss of white matter is reported in both the middle cerebellar peduncles and on the outer edge of the pons (Abele et al. 2007).
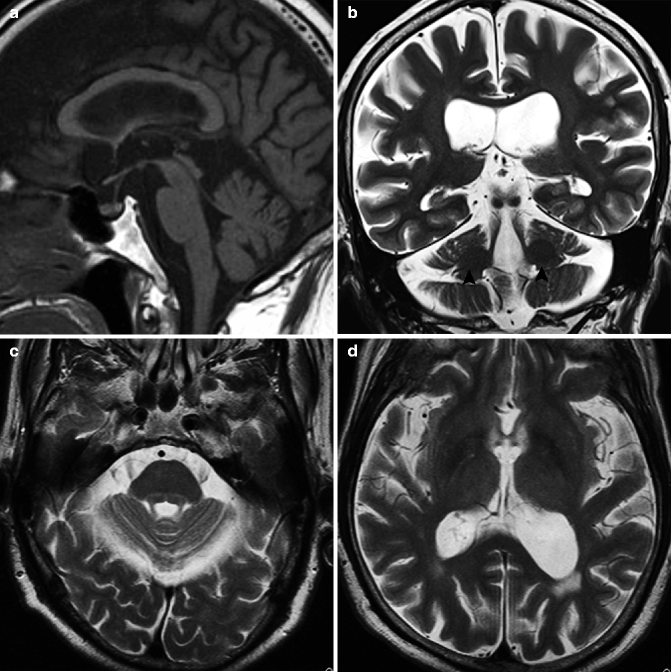
Fig. 25.10
Cortical cerebellar atrophy. 77-year-old man with wobbliness. (a) T1-weighted image, sagittal; (b) T2-weighted image, coronal; (c, d) T2-weighted image, axial. Atrophy in cerebellar cortical gray matter can be seen (b). There is no sign of atrophy of transverse pontine fibers or atrophy of brainstem (a, c). Middle cerebellar peduncles (arrowheads) are also preserved in this case (b)
25.4.2 Autosomal Dominant Cerebellar Atrophy
Hereditary SCD includes autosomal dominant disorders such as dentatorubral-pallidoluysian atrophy (DRPLA) and spinocerebellar ataxia (SCA) and autosomal recessive disorder. Autosomal recessive disorder includes Friedreich ataxia and early-onset ataxia with ocular motor apraxia and hypoalbuminemia (EAOH). Recently, the specific gene responsible for the anomaly has been identified for a variety of disorders, and the classification of the diseases have become clearer. For example, there are 31 types of genetic disorders identified for spinocerebellar ataxia (SCA). SCA is a genetically heterogeneous group of cerebellar degenerative disorders, for which symptoms include progressive gait instability, incoordination, and dysarthria due to degeneration of the cerebellum or its connections. The characteristic clinical findings of hereditary SCD is anticipation, which is a phenomenon in which the onset age of the disease becomes earlier and the symptoms become more severe in successive generations.
25.4.2.1 Spinocerebellar Ataxia Type 1 (SCA1)
Spinocerebellar ataxia type 1 (SCA1) is the first disorder for which the responsible gene anomaly has been identified (Orr et al. 1993). This disorder is one of the polyglutamine disorders which are caused by a polyglutamine expansion in ataxin-1 due to the expansion of an unstable CAG repeat (Zoghbi and Orr 2000). The onset of symptoms is in the fourth to fifth decade of life, and symptoms include slow-growing cerebellar ataxia, oculomotor palsy, and pyramidal symptoms.
The pathological changes in this disease can be seen in the inferior olivary nucleus, pontine nucleus, and cerebellar cortex, with mild neuronal loss and gliosis (Matilla-Duenas et al. 2008). The external segment of the globus pallidus also shows degeneration, while the striatum is preserved. There may be degeneration in the dentate nucleus, oculomotor nuclei, and medial longitudinal fasciculus. The major imaging findings in this disorder are seen in the cerebellum and brainstem as they atrophy (Fig. 25.11). The degeneration of the transverse pontine fibers can also be seen.
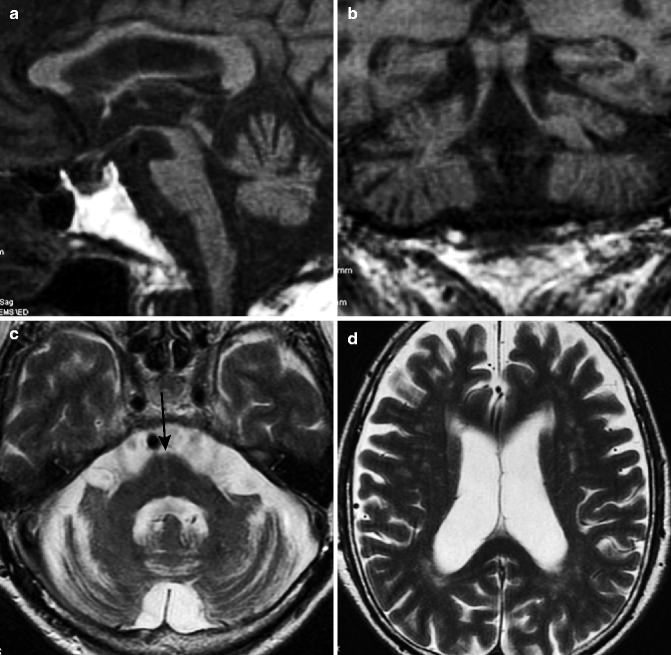
Fig. 25.11
Spinocerebellar ataxia type 1 (SCA1). 61-year-old man with dysarthria and gait disturbance. (a) T1-weighted image, sagittal; (b) T1-weighted image, coronal; (c, d) T2-weighted image, axial. Severe atrophy in cerebellum and brainstem can be seen (a–c). Dilation of fourth ventricle is visible (a, c). Middle cerebellar peduncle also shows atrophy (b). On T2-weighted image, hyperintensity can be seen in midline of pons (arrow), which suggests degeneration of transverse pontine fibers (c). White matter of cerebral hemisphere also shows slight volume loss (d) (By courtesy of Professor Shoki Takahashi, Department of Radiology, Tohoku University)
25.4.2.2 Spinocerebellar Ataxia Type 2 (SCA2)
Spinocerebellar ataxia type 2 is autosomal dominant and the spinocerebellar degeneration is caused by mutations in the ataxin-2 gene on chromosome 12q24.1. The mutation is the expansion of the CAG trinucleotide repeat. Degeneration of the inferior olivary nuclei, pontine nuclei, and cerebellar cortex without degeneration of the dentate nuclei is the major pathological change associated with this disorder. The onset age is widely distributed. The symptoms include slowly progressing cerebellar ataxia, weakened tendon reflex, slow saccades, oculomotor palsy, involuntary movement, and loss of vibration sensation.
On images, atrophy of the cerebellar hemispheres with white matter volume loss can be seen. Atrophy of the cerebral peduncle, cerebellar cortex, pontocerebellar tract, and olive are also apparent. There are some cases reported with linear midline hyperintensity or hot cross bun sign at the ventral pons like MSA-C (Lee et al. 2009).
25.4.2.3 Spinocerebellar Ataxia Type 3 (SCA3: Machado-Joseph Disease)
Spinocerebellar ataxia type 3, also called Machado-Joseph disease, is one of the autosomal dominant spinocerebellar degenerative diseases caused by a mutation in the MJD-1 gene on chromosome 14q32. The mutation is the expansion of the CAG trinucleotide repeat. The severity of symptoms depends on the number of CAG repeats. Earlier onset in successive generations has been demonstrated and is known as anticipation. The organs that are impaired in this disorder show wide variation, including the substantia nigra, dentate nucleus, red nucleus, pontine nuclei, other cranial nerve nuclei, Clarke column, and the anterior horn cells of the spinal cord. Thus, atrophy of the pons and superior cerebellar peduncles, and widening of the fourth ventricle may accompany this disorder. The inferior olivary nucleus is usually preserved. The onset age for SCA3 is widely distributed. In the early stage of the disease, progressive motor disorders can be seen, including cerebellar ataxia, pyramidal tract symptoms, oculomotor disorder, bulging eyes, dystonia, and athetosis.
On images, cerebellar atrophy is the most common finding even in the early stage of the disease (Fig. 25.12). The cerebellar atrophy is most evident in the vermis and white matter, while the cerebellar cortex is relatively preserved. In the pons, the atrophy can be seen both in the ventral region and tegmentum. Atrophy of the pontine tegmentum is more prominent; thus, the dorsal area of the pons looks concave in sagittal images. There may be degeneration of transverse pontine fibers, which show linear high signal on T2-weighted images in the midline of the ventral pons, which resembles MSA-C, but these symptoms of SCA3 are relatively mild compared to MSA-C. The midbrain, globus pallidus, and frontal lobes also show atrophy in the later stages of the disease. Abnormal signal in the globus pallidus has also been reported (Shirai et al. 2007).
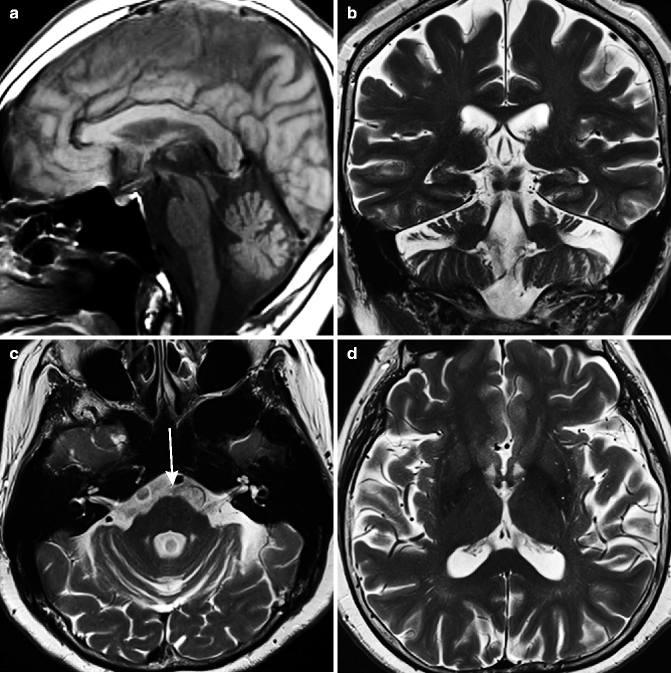
Fig. 25.12
Spinocerebellar ataxia type 3 (SCA3: Machado-Joseph disease). 67-year-old woman with cerebellar ataxia. (a) T1-weighted image, sagittal; (b) T2-weighted image coronal; (c, d) T2-weighted image, axial. Marked cerebellar atrophy can be seen in vermis and white matter (a–c). Cerebellar cortex appears to be preserved. Pontine atrophy can be seen both in ventral region and tegmentum; however, atrophy of pontine tegmentum is more prominent (a, c). Linear slightly high signal on T2-weighted image in midline of ventral pons is apparent (arrow), suggesting degeneration of transverse pontine fibers (c)
25.4.2.4 Spinocerebellar Ataxia Type 6 (SCA6)
The symptoms of this disorder are almost pure cerebellar ataxia. SCA6 is another of the CAG repeat diseases; however, anticipation has not been reported. The mean age of onset ranges from 43 to 52 years. In most cases, the first symptom is ataxic gait. Almost all cases show cerebellar ataxia, and dysarthria or nystagmus occur in most of the cases. The prognosis for SCA6 is rather favorable. Pathological changes associated with this disorder include neuronal loss of Purkinje cells and granular cells of the cerebellum and loss of neurons in the inferior olivary nucleus.
On the image, atrophy of the cerebellar hemisphere can be seen, especially in the upper region of the hemisphere (Fig. 25.13). Significant superior vermian atrophy has also been reported (Butteriss et al. 2005). The brainstem is preserved, and atrophy or abnormal signals related to degeneration of the pontine transverse fibers cannot be seen.
Get Clinical Tree app for offline access
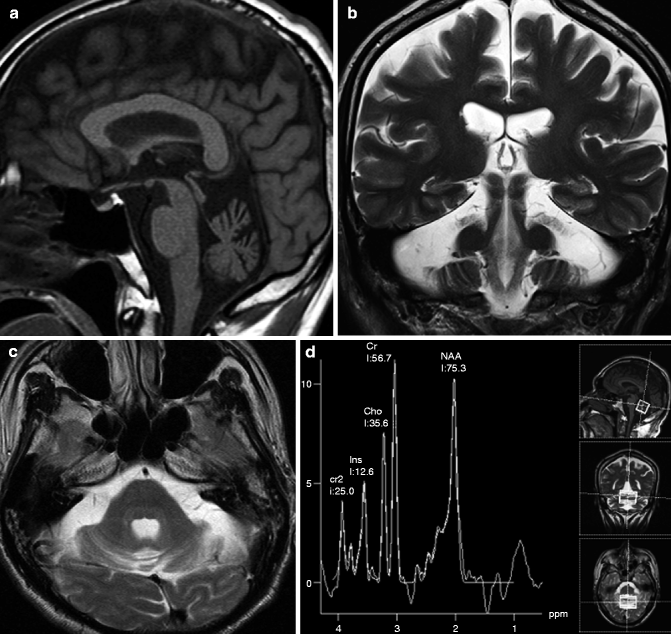
Fig. 25.13
Spinocerebellar ataxia type 6 (SCA6). 52-year-old man with wobbliness. (a) T1-weighted image, sagittal; (b) T2-weighted image, coronal; (c) T2-weighted image, axial; (d) MR spectroscopy. Atrophy of cerebral hemisphere can be seen especially in dorsal part of hemisphere (b). Superior part of vermis also shows atrophy (a). Volumes of middle cerebellar peduncle and brainstem are preserved (a, c




Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
