Musculoskeletal Disorders Due to Endocrinopathy, Metabolic Derangement, and Arthropathy
Ricardo Restrepo
Edward Y. Lee
Paul S. Babyn
Hadeel M. Seif El Dein
Bjorn Lundin
Andrea S. Doria
INTRODUCTION
The pediatric skeleton is an important dynamic system. Bone plays many relevant physiologic roles such as providing hematopoiesis and structural support while helping maintain cellular and chemical blood homeostasis. Endocrine, metabolic, and arthritic diseases often affect the pediatric skeleton as normal bone development in children requires intense anabolic activity. Disorders of hormones, growth and humoral factors, vitamins, and minerals may all lead to changes visible to the radiologist. This chapter reviews the up-to-date imaging techniques and characteristic imaging findings of those bone disorders that occur because of underlying endocrine and metabolic abnormalities as well as arthropathies that can be encountered in the pediatric population.
IMAGING TECHNIQUES
Radiography
Conventional radiography remains the most commonly used initial imaging modality for the evaluation of bone and joint disorders in pediatric patients with clinically suspected endocrine, metabolic, and arthropathic abnormalities. Radiographs are readily available, easy to obtain, and low cost and have minimal radiation exposure.1 The appearance of the epiphyses, metaphyses, and physes as well as bone density and shape must be carefully assessed.
A typical radiographic series for evaluation of suspected endocrine and metabolic abnormalities includes anterior-posterior views of the wrists/hands and knees, as bone changes are most conspicuous in areas of active growth. Bilateral views are recommended to confirm systemic involvement. Bone age can also be determined on the hand radiograph (or the knee radiograph in children who are younger than 1 year of age) in order to determine delayed maturation.2 A skeletal survey may be useful in the evaluation of the bones of children with systemic disorders (i.e., metabolic, endocrine) or skeletal dysplasias or when nonaccidental trauma is suspected.
The evaluation of children with suspected osteochondroses also begins with radiographs of the affected area. In children, radiographs of the contralateral side may be helpful to confirm subtle abnormalities and exclude normal developmental variants.1 There may be the need for two or more views in the imaging assessment of certain osteochondroses such as slipped capital femoral epiphysis or osteochondritis dissecans (OCD) of the knee as described later in this chapter. In children with juvenile idiopathic arthritis (JIA), radiographs are useful at the time of the initial diagnosis to exclude other diagnostic pathologies and in the evaluation of complications occasionally seen late in the disease. In JIA patients, radiographs provide an overall appearance of the disease and information regarding bone mineralization. On the other hand, radiographs have poor sensitivity in detecting early changes of JIA and evaluating JIA activity because of their poor capability of assessment of soft tissues and cartilage.3
Ultrasound
Gray-scale as well as color and power Doppler ultrasound (US) may play an important role in evaluating children with underlying endocrine, metabolic, and arthropathic abnormalities. Without requiring sedation or exposing children to ionizing radiation, US can provide information regarding the synovium, marginal articular cartilage, joint effusions, and periarticular soft tissues. In skeletally immature pediatric patients, because of the larger extent of cartilaginous structures, US can be used to evaluate the unossified and superficial portions of bones.
High-frequency (7.5 to 15 MHz) linear array transducers, which can provide excellent spatial resolution of superficial musculoskeletal structures and pathology, are typically used in young children. However, lower-frequency curved or vector US transducers, which can provide increased soft tissue penetration, may be needed in older pediatric patients with larger body habitus. Color and power Doppler US imaging, which can provide information regarding vascularity and blood flow, is useful in the assessment of underlying inflammation. For example, in children with JIA, US is helpful in assessing the extent and severity of joint disease, including early erosions, and in monitoring treatment effectiveness and excluding subclinical disease.3,4
Computed Tomography
Computed tomography (CT), which can provide superb visualization of osseous structures, can be a useful imaging modality in children with underlying endocrine and metabolic abnormalities as well as arthropathies. However, as it is associated with ionizing radiation, judicious use is necessary. Multidetector CT (MDCT) with two-dimensional (2D) and three-dimensional (3D) reconstruction capabilities is particularly helpful for assessment of complex anatomic regions such as the face, rib cage, and hip.1 Such 2D- and 3D-reconstructed CT images can be used as preoperative imaging tool to guide the orthopedic surgeon’s interventions in pediatric patients with osseous deformities secondary to Legg-Calve-Perthes (LCP) and Blount disease.5,6
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) provides the most comprehensive evaluation of musculoskeletal structures in pediatric patients clinically suspected of having underlying endocrine, metabolic, and arthropathic abnormalities. Its multiplanar capabilities and the ability to evaluate bone marrow as well as the cortex can be useful in the imaging evaluation of pediatric patients with inflammatory arthropathies and systemic diseases such as sickle cell disease (SCD) and Gaucher disease.3,7,8 Additionally, MRI is currently the best imaging modality to evaluate cartilage morphology in pediatric patients with inflammatory arthropathies such as JIA or osteochondrosis such as OCD.4 Disadvantages of MRI include its higher cost and lower availability in comparison to the other imaging modalities, longer duration of the study, and need for intravenous contrast or sedation in young patients. In many cases, because of the long scanning time, MRI only allows the evaluation of a single joint at a time. However, more recent MRI techniques, such as whole-body MRI and parallel imaging, can now allow a relatively fast evaluation of the entire body for systemic diseases such as spondyloarthropathies and childhood inflammatory myopathies.9,10
Nuclear Medicine
Bone scintigraphy is a modality that detects the distribution and uptake of a radioactive agent injected into the vascular system throughout the body. The main advantage of bone scintigraphy is its ability to evaluate the entire skeleton at once, which is useful in pediatric patients with underlying systemic and metabolic disorders. It allows the functional evaluation of the soft tissues, cortical bone, and bone marrow. Bone scintigraphy is a sensitive but not specific imaging modality that can help confirm the presence of disease in the early stages. This is critical in pediatric patients with certain osteochondroses, such as early LCP disease and slipped capital femoral epiphysis, that can lead to substantial disability if not accurately diagnosed in a timely fashion. In systemic disorders such as SCD, bone scintigraphy can provide an overall appearance of the entire skeleton. In JIA, bone scintigraphy currently plays no routine role. Disadvantages of bone scintigraphy include its use of radiation, limited spatial resolution which may be improved with SPECT imaging, and the possible need of sedation.1
SPECTRUM OF DISORDERS
Musculoskeletal Disorders Due to Endocrinopathy and Metabolic Derangement
Rickets
Rickets results from a disruption of the orderly development and mineralization of the growth plates.11 Rickets affects the physes by two main mechanisms: (1) deficient mineralization of cartilage and osteoid due to low calcium or phosphate and (2) retarded endochondral ossification, which causes excessive accumulation of cartilage in the physis, growth failure, and skeletal deformities.12
The causes of rickets are varied and can be divided into two main groups: calcipenic or phosphopenic, depending on whether the initial problem results in insufficient calcium absorption or excessive phosphate excretion, respectively. Causes of calcipenic rickets include vitamin D abnormalities and calcium deficiency, certain medications such as anticonvulsants (e.g., phenytoin), malabsorption syndromes, and obesity. The latter is due to calcium sequestration in excessive adipose tissue. Dietary calcium deficiency is uncommon in most of the world.2,12 Rickets can be associated with prematurity and is related to a combination of nutritional, metabolic, and iatrogenic factors, usually manifesting around 12 weeks of age or later in infants who are born younger than 32 weeks of gestational age or weighing <1,500 g (Fig. 25.1).2,11,13 Phosphopenic rickets is most commonly caused by hereditary or acquired disorders of renal tubular phosphate wasting, the most frequent of which is X-linked hypophosphatemia (familial vitamin D-resistant rickets) (Schematic D).12
 CKDMBD, chronic kidney disease-mineral bone disorder; HP, hypoparathyroidism; MPS, mucopolysaccharidosis; PHP, pseudohypoparathyroidism; PPHP, pseudopseudohypoparathyroidism; PHPT, primary hyperparathyroidism; SCD, sickle cell disease. |
 |
 FIGURE 25.1 A 5-month-old boy with history of severe prematurity and rickets. Frontal chest radiograph shows severely diffuse osteopenia and a rosary appearance of the costochondral junctions (arrows). |
Radiographs play an important role in the diagnosis of rickets and in the evaluation of treatment response. A typical metabolic radiographic series includes views of the hand and knees, as rachitic changes are more conspicuous in areas of most active growth. Bilateral views are recommended to confirm a systemic involvement. Bone age can also be determined to assess delayed maturation.2,14 Nonspecific radiographic changes of rickets include diffuse demineralization (osteopenia), coarsening of the trabecular pattern, and the lack of growth that precedes physeal changes. The more typical changes of rickets affect the growth plate, with physeal widening being the first sign to appear. As the disease progresses, the metaphyseal margin, more specifically the zone of provisional calcification, is lost, and the physis widens. Metaphyseal fraying and cupping occur in the affected long bones (Fig. 25.2 and Schematic B). Epiphyses progressively become more lucent until their margins are lost. In the shafts, radiographic findings of rickets include subperiosteal resorption, cortical thinning, intracortical tunneling, and endosteal resorption. Additional diaphyseal findings include insufficiency fractures, periosteal reaction, and bowing deformity. Bowing is more pronounced in the lower extremities with weight bearing.11,14,15
With healing, radiographic changes are seen by 2 to 3 months, with radiographic changes lagging clinical and laboratory parameters by a few weeks. The earliest radiographic finding of healing rickets is the revisualization of the zone of provisional calcification as an opaque line separated from the rest of the shaft with subsequent mineralization of the underlying physeal cartilage. Periosteal reaction can also be seen in the healing phase. Bowing deformities may persist for a long time.15
In hypophosphatemic rickets (Fig. 25.3), bone mineralization is better preserved than it is in vitamin D-deficient rickets. Major clinical differences between these two types of rickets include short stature and bowing, particularly of the lower extremities. Bowing is more frequently present in hypophosphatemic rickets. Insufficiency fractures and looser zones are also more commonly seen in hypophosphatemic rickets than in the vitamin D-deficient type of rickets.11,15
 FIGURE 25.2 A 14-month-old boy with vitamin D deficiency rickets. Frontal tibia and fibula radiograph shows diffuse osteopenia, metaphyseal splaying, fraying, and cupping of the distal femur, tibia (arrow), and fibula. |
Hyperparathyroidism
Primary hyperparathyroidism (PHPT) is one of the most common endocrine disorders, with a prevalence of 0.1% to 0.4% and an incidence that increases with age, to peak in the sixth decade.17 PHPT is uncommon in adolescents and is rare in children, having a prevalence of 3% to 5% in Western countries.18 PHPT is defined as hypercalcemia secondary to the overproduction of parathormone (PTH) by one or more parathyroid glands in which the negative feedback to regulate calcium is lost, resulting in increased renal absorption of calcium, increased synthesis of vitamin D, phosphaturia, and increased bone resorption.17 The classic symptoms include bone and abdominal pain, psychiatric symptoms, and fatigue. Affected pediatric patients also often present with renal stones. In comparison to adults with PHPT, affected pediatric patients present with a higher frequency of symptoms and more end organ damage.17,19,20
In the pediatric population, PHPT can be divided into two groups by age of onset: neonatal and childhood types.
Neonatal HPT has an autosomal recessive inheritance or may be due to maternal hypothyroidism. Neonatal HPT may present in newborns and infants with life-threatening hypercalcemia due to parathyroid hyperplasia.21,22 In older children and adolescents, most cases of PHPT are sporadic because of a parathyroid adenoma followed by glandular hyperplasia. Some of these pediatric patients have multiple endocrine neoplasia syndrome or familial isolated PHPT.17,18,20
Neonatal HPT has an autosomal recessive inheritance or may be due to maternal hypothyroidism. Neonatal HPT may present in newborns and infants with life-threatening hypercalcemia due to parathyroid hyperplasia.21,22 In older children and adolescents, most cases of PHPT are sporadic because of a parathyroid adenoma followed by glandular hyperplasia. Some of these pediatric patients have multiple endocrine neoplasia syndrome or familial isolated PHPT.17,18,20
 FIGURE 25.3 Oncogenic hypophosphatemic rickets in an 11-year-old boy. Bony spicules are markedly diminished in size and number (left; hematoxylin and eosin, original magnification, 40×). In this case, the phosphate dysregulation resolved after resection of a phosphaturic mesenchymal tumor occurring in the tibia (right; hematoxylin and eosin, original magnification, 200×). |
 FIGURE 25.4 Primary hyperparathyroidism in a 15-year-old boy with a parathyroid adenoma. A: Frontal hand radiograph reveals coarsening of the trabecular pattern and signs of bone resorption such as cortical tunneling (straight arrows) and acroosteolysis (curved arrow). B: Frontal skull radiograph shows the classic “salt and pepper”-appearing skull (arrows) indicative of bone resorption. |
The imaging modality of choice for evaluating bone involvement in children with PHPT is radiography. Typical radiographic findings include osteopenia, coarsening of the trabeculae, subperiosteal bone resorption, and cortical tunneling (Fig. 25.4A). Subligamentous bone resorption is also common, more typical in the distal femur and proximal tibia. Trabecular bone resorption may occur throughout the skeleton, including the skull, where it produces the “salt and pepper” appearance (Fig. 25.4B and Schematic A). Metastatic soft tissue and visceral calcifications, chondrocalcinosis, slipped capital femoral epiphyses, and bone deformities (i.e., genu valgum) may occur as well.15,21
Typically, PHPT in the pediatric population has been managed with bilateral neck exploration and biopsy of the parathyroid glands. In cases of confirmed single parathyroid adenoma, exploration and excision of the single affected gland can be performed.20
Chronic Kidney Disease-Mineral Bone Disorder
Chronic kidney disease-mineral bone disorder (CKDMBD), formerly known as renal osteodystrophy (ROD), refers to the characteristic musculoskeletal manifestations of hyperparathyroidism secondary to chronic renal failure.2,15 Both glomerular and tubular dysfunctions contribute to the development of CKDMBD. Tubular dysfunction causes hypocalcemia because of deficient vitamin D synthesis, whereas impaired glomerular function causes phosphorus retention. This results in hypocalcemia that triggers hyperparathyroidism in an effort to restore calcium levels. Secondary hyperparathyroidism is the major metabolic abnormality in ROD. As the disease progresses, rickets and osteomalacia develop.2,15,23
Radiographic changes of hyperparathyroidism stem from increased osteoclastic bone resorption. Classic radiographic features include subperiosteal, endosteal, and subligamentous resorption as well as osteopenia with coarsening of the trabecular pattern. Subperiosteal resorption is most frequently seen in the middle phalanges of the hand (Fig. 25.5A and Schematic B), distal clavicle (Fig. 25.5B), and distal femur and proximal tibia. Endosteal resorption, also known as cortical tunneling, is seen as a lacy pattern of the inner cortex of the bone. Acroosteolysis, which represents terminal tuft bone resorption, is also a manifestation of CKDMBD (Fig. 25.5A). Subchondral resorption that is adjacent to the physeal cartilage, the so-called osteitis fibrosa, can also be seen causing lucency at the edge of the physis, simulating rickets and predisposing to epiphyseal slippage. Osteosclerosis also is a common manifestation that causes diffuse chalky bone density, which in the spine affects the vertebral end plates, producing the rugger jersey spine.2,11,15,24 A real measurement of bone mineral density by dual x-ray absorptiometry is currently available to assess bone mass in the pediatric population.25 Soft tissue and visceral calcification are also a manifestation of CKDMBD because of the deposition of calcium salts.26
Histologically, the bone in renal bone disease shows prominent osteoclastic and osteoblastic activity, increased woven bone, and marrow fibrosis that begin as paratrabecular (Fig. 25.5C) and progresses to diffuse (osteitis fibrosa). Control of bone and mineral homeostasis is very challenging in children, with most of the affected patients experiencing signs of hyperparathyroidism to different extents.
The treatment consists of dietary counseling, ergocalciferol and cholecalciferol supplementation, and the use of calcium-free phosphate binders.23
Tertiary Hyperparathyroidism
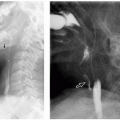
Tertiary hyperparathyroidism (THPT) is rare in children and adolescents, and relatively little is currently known about its radiologic findings. The diagnosis of this entity is mainly based on laboratory findings.27 The boundary between secondary (nonautonomic function) and tertiary (autonomic function) forms of hyperparathyroidism is sometimes blurry with radiologic findings of THPT representing more advanced stages of ROD (Fig. 25.6). This entity is a manifestation of the uremic state of CKDMBD. Although the characteristic autonomic function of the parathyroid glands in THPT may resemble the metabolic activity of an adenoma, this pathologic entity is in fact rare in THPT. Instead, most cases (94%) of THPT present with marked glandular hyperplasia.27,28
99mTc sestamibi (MIBI) scintigraphy is very sensitive and accurate for preoperative localization of parathyroid lesions in patients with PHPT and has a fair sensitivity and accuracy to detect all abnormal parathyroid glands in multiple hyperplasia.29 Nevertheless, MIBI is an important diagnostic technique for identifying ectopic parathyroid glands in the mediastinal and thymic regions in patients with THPT.30
Hypoparathyroidism, Pseudohypoparathyroidism, and Pseudopseudohypoparathyroidism
Hypoparathyroidism (HP) is an abnormality of the calcium metabolism characterized by low serum levels of PTH in spite of hypocalcemia and secondary hyperphosphatemia.31 Primary idiopathic HP can be familial or sporadic. It can be associated with several syndromes, including DiGeorge syndrome, which is characterized by congenital absence of the parathyroid glands. Other causes of HP are autoimmune disorders and postsurgical resection. Pseudohypoparathyroidism (PHP) is a hereditary disease characterized by similar findings of HP; however, the parathyroid glands are normal with elevated PTH due to end organ resistance to PTH. Pseudopseudohypoparathyroidism (PPHP) is the normocalcemic form of PHP.21,31,32
The musculoskeletal manifestation of HP can be evaluated using radiographs. The most common radiographic feature of HP that occurs in ˜20% of affected patients is generalized or focal osteosclerosis, particularly in the calvarium, pelvis, and proximal femora.21 The face and the skull are areas that are frequently involved in affected pediatric patients. Radiographic findings include thickening of the cranial vault and facial bones, sutural diastasis in cases with associated increased intracranial pressure, and abnormal tooth development with delayed eruption or supernumerary teeth. Other manifestations include premature epiphyseal fusion, dense metaphyseal bands, and calcifications in the soft tissue, tendons, and spinal ligaments.11,21
 FIGURE 25.5 A 13-year-old girl with renal osteodystrophy from underlying end-stage renal disease. A: Frontal hand radiograph shows diffuse osteopenia and signs of bone resorption such as cortical tunneling (straight arrows) and acrosteolysis with tuft resorption (curved arrows). B: Frontal chest radiograph demonstrates resorption of the distal clavicles and scapular tips (straight arrows) and widening of the costochondral junctions (curved arrows). Note calcified thrombus of the superior vena cava due to previous dialysis catheters (arrowhead). C: Chronic kidney disease-mineral bone disorder in an 11-year-old girl with renal failure. A posterior iliac crest biopsy shows marrow fibrosis and decreased hematopoiesis (hematoxylin and eosin, original magnification, 400×). |
A typical radiographic finding in PHP and PPHP is the shortening of one or more of the metacarpals and metatarsals due to the premature fusion of the physis, most commonly affecting the fourth and fifth rays. This finding occurs more frequently in PPHP and most commonly involves the hands (Fig. 25.7 and Schematic B). Other skeletal features of PHP and PPHP include diaphyseal exostoses, coxa valga or vara (Schematic I), osseous bowing, cone epiphyses, and accelerated skeletal maturation. Soft tissue calcification similar to HP can also be seen.21
The majority of cases of pediatric PHP are well controlled with the administration of calcium and vitamin D analogues. For nonresponding cases, PTH replacement therapy can be used; however, the use of recombinant human PTH is contraindicated in children because of the increased risk of osteosarcoma.31,32
Hypophosphatasia
Hypophosphatasia is characterized by a defective bone mineralization caused by insufficient activity of tissue-nonspecific alkaline phosphatase. This eventually leads to the excessive accumulation of inorganic pyrophosphate, resulting in deficient bone mineralization. The four main types of hypophosphatasia are perinatal, infantile, childhood, and adult types. Similar to rickets, the diagnosis of hypophosphatasia can be made based on radiographs. In hypophosphatasia, there is lack of mineralization of parts of the skeleton which is more severe than the mineralization defect noted in osteogenesis imperfecta. The infantile and childhood forms resemble rickets with less uniform and patchier metaphyseal involvement. The adult form simulates osteomalacia with a coarse trabecular pattern, metatarsal stress fractures, and Looser zones.33
 FIGURE 25.6 A 17-year-old girl with granulomatosis and polyangiitis (previously known as Wegener granulomatosis) and chronic renal failure diagnosed with tertiary hyperparathyroidism on the basis of laboratory findings. Lateral radiograph of the thoracolumbar spine shows sclerosis of the vertebral endplates (arrowheads) with multilevel intervertebral disk calcification foci (arrows). |
No effective treatment currently exists for hypophosphatasia. There is only treatment for specific symptoms and supportive measures.33
Scurvy
Scurvy is a disease that results from a physiologic deficiency of vitamin C, now rarely seen in the developed world. Humans are dependent on the exogenous intake of vitamin C that is present in vegetables and fruits. Vitamin C is also present in breast milk.34,35,36 In children, scurvy is more common in infants who are fed with pasteurized or boiled milk, as heat disrupts the vitamin, and it also is more common in preschool children who have an inadequate intake of vegetables and fruits. Infantile scurvy usually develops after 6 months of age because of the storage of vitamin C built up in utero and during the first few months in life through breast milk.35 Affected young children present with failure to thrive and gastrointestinal (GI) symptoms. As the disease progresses, affected pediatric patients develop gum bleeding and bone changes. Skin manifestations and bleeding are more common in older children and adults.35
The diagnosis of scurvy is made based on serum levels of vitamin C and on the radiographic skeletal manifestations that include osteoporosis and abnormalities in the areas of active endochondral growth, such as at the end of the long bones. Biopsy may occasionally be pursued (Fig. 25.8A). Extensive resorption of cortical and cancellous bone predisposes these areas to fracture (Fig. 25.8A) that often display excessive callus during the healing phase. The Trummerfeld zone is a disrupted metaphyseal zone of osteoporosis that can result in peripheral metaphyseal excrescences if there is associated healing (Fig. 25.8B and Schematic C) and osseous malalignment at the metaphyseal-diaphyseal junction due to physeal separation. The Wimberger ring sign is characteristic of the disease and consists of a radiodense line at the edge of an ossification center that corresponds to the normal mineralized cartilaginous zone of provisional calcification, which contrasts more with the demineralized remainder of the center (Fig. 25.8B). Alternating dense and lucent metaphyseal lines can be present along with hemarthrosis and subperiosteal hemorrhage, which is seen as periosteal reaction along the long bones.11,35,37 More recently, MRI has been used to evaluate scurvy in pediatric patients with autism.38
 FIGURE 25.7 A 9-year-old boy with pseudopseudohypoparathyroidsim. Frontal hand radiograph shows a short fourth metacarpal (asterisk). |
The treatment of scurvy includes vitamin C supplementation, which typically leads to a rapid resolution of the symptoms. For infantile scurvy, avoiding pasteurized or boiled milk and providing vitamin supplementation to the lactating mother is recommended in order to increase the vitamin’s level in the breast milk.36 Radiographically, cortical thickening, dense metaphyseal lines, exaggerated subperiosteal bone formation, and osseous realignment are signs of healing.11,35
 FIGURE 25.8 A: Scurvy due to vitamin C deficiency in an 11-year-old boy with a history of autism and escalating food aversions. An iliac crest biopsy shows a paratrabecular spindle cell proliferation (hematoxylin and eosin, original magnification, 400×). B: Scurvy in a 6-year-old boy. Frontal knee radiograph shows a linear metaphyseal lucency with an associated fracture and a focal bony excrescence indicating early healing (Trummerfeld zone) (curved arrow). Significant osteopenia, more conspicuous in the epiphysis with a peripheral sclerotic line (Wimberger ring sign), is seen (asterisks). |
Heavy Metal Poisoning
Heavy metals (HM) are defined as those metals that have a specific density of more than 5 g/cm3, such as lead, mercury, aluminum, arsenic, cadmium, and nickel. They are widely distributed in the earth’s crust, and most are normally present at very low concentrations in the body. HM are toxic because of potential cumulative deleterious effects that can cause chronic degenerative changes, especially in the nervous system, liver, kidney, and bones.39,40
The mechanisms of toxicity of most of these agents are well known and include enzymatic inhibition, impaired antioxidant metabolism, oxidative stress, and free-radical formation.39 In modern life, most cases of HM poisoning occur in adults because of occupational hazards; however, lead poisoning still raises serious concern in children.40,41,42 Lead has industrial use but no physiologic use; therefore, any evidence of lead in the body should be considered contamination. In children, the mechanism of poisoning is accidental ingestion and most frequently occurs in toddlers. The primary route of lead exposure is through the GI tract, where lead is taken up at calcium absorption sites, which are very active at times of rapid growth. Being an inorganic compound, it is poorly absorbed through the GI tract. Chronic exposure to very small amounts of lead is necessary to cause untoward effects. Lead-based paints in households and toys are common causes of lead intoxication in children.40,42 The most common childhood presentation of lead poisoning (plumbism) is central neurotoxicity. Other symptoms of lead toxicity include anemia, anorexia, abdominal pain, vomiting, and growth delay.40,41,42
The diagnosis of lead intoxication, as of any other heavy metal toxicity effect, is made by measuring the blood levels of the metal in question. Pediatric patients in high-risk areas should be screened every 1 to 2 years. The skeletal manifestations of lead poisoning can be evaluated using radiographs. Typical features include dense transverse metaphyseal lines due to the increased deposition of calcium along with lead, most prominently around the knee. Dense bands in the femur and tibia can be seen in normal children; thus, the presence of fibular bands supports the diagnosis (Fig. 25.9). Radiodense bands can be seen in other sites of the axial and appendicular skeleton, including the spine. If lead exposure is interrupted, these dense bands slowly migrate to the diaphysis and disappear in a few years. Dense metaphyseal bands are not characteristic of lead poisoning because they can be seen with other types of HM intoxication, scurvy, rickets, and treated leukemia. Abnormal tubular bone remodeling, most pronounced in the distal femur with metaphyseal widening as well as sutural widening in the skull due to increased intracranial pressure, can be seen.11
 FIGURE 25.9 Lead poisoning in a toddler boy. Frontal tibia and fibula radiograph shows a typical dense metaphyseal band (lead band) (arrows) around the knee and ankle. The proximal fibular involvement is more supportive of the diagnosis of lead poisoning. |
Sickle Cell Disease
Bone involvement is the most common manifestation of SCD, both acutely and chronically. Acute manifestations of SCD include painful vasoocclusive crisis, osteomyelitis, bone marrow infarcts, stress fractures, and vertebral collapse (“fish-type” or “H-shaped” vertebrae; Fig. 25.10 and Schematic F). Chronic manifestations are mainly related to osteonecrosis and secondary bone collapse and deformity.44 In this chapter, the vasoocclusive crisis and bone infarcts from SCD are discussed.
Vasoocclusive crisis affects virtually all pediatric patients with SCD, often beginning in late infancy and recurring throughout their lives. The pathogenesis of vasoocclusive crisis relates to microvascular occlusion due to the abnormally deformed red cells that may be seen in any organ but occur more frequently in the bone marrow. These microvascular occlusions result in bone marrow infarction, typically in the medullary cavity or epiphyses.44,45
 FIGURE 25.10 Sickle cell disease in a 15-year-old boy. The vertebral column shows large areas of medullary pallor (infarcts) as well as extensive vertebral body collapse. |
Clinically, affected pediatric patients complain of localized or multifocal intense pain, edema and erythema with or without fever and leukocytosis.44,45 Dactylitis is a fairly classic presentation of vasoocclusive crisis in younger children who are typically between 1 and 2 years; it involves the small bones of the hands and feet, which contain hematopoietic marrow (Fig. 25.11). Affected children present with a swollen and painful digit.7,44,46 Osteonecrosis occurs when the vasoocclusion results in infarction of the epiphyses and may involve the articular surfaces.44,45
The diagnosis of a painful crisis is predominantly clinical. Radiographs are of little value, as the acute changes are usually not seen. Bone scintigraphy or MRI is the imaging modalities of choice during an acute pain crisis. Bone scintigraphy is helpful in assessing multifocal involvement showing increased uptake in areas of acute bone marrow infarction.44 More recently, whole-body MRI with diffusion-weighted imaging (DWI) sequences has been used successfully not only to evaluate the extent of disease but also to evaluate the acuity of the infarcts.47,48 In ischemia, DWI shows increased diffusion within the first hours of the insult and remains elevated during the healing process.47 When the involvement is localized, MRI may be more helpful, as it is very sensitive in detecting bone marrow and soft tissue abnormalities. In the acute setting, bone infarcts are hyperintense on fluid-sensitive sequences and hypointense on T1-weighted MR images. On T1-weighted MR images with fat saturation before contrast, acute lesions tend to be hyperintense and display no enhancement after contrast. When epiphyseal in location, infarcts can be associated with joint effusion. Adjacent soft tissue edema and stranding are findings that support a more acute process.
The more chronic infarcts tend to be predominantly hypointense with a peripheral bright halo on fluid-sensitive sequences and hypointense with fatty elements on T1-weighted MR images (Fig. 25.12A). SCD-related bone marrow infarcts tend to be larger than those from other etiologies, reflecting a more systemic disease. More chronic infarcts can be seen on radiographs as serpiginous lytic lesions with a peripheral sclerotic line (Schematic C). On radiographs, diffuse or patchy osteosclerosis is also present (Fig. 25.12B). In the epiphyses, a subchondral crescentic lucency is fairly characteristic on radiographs (Schematic F). On MRI, the lucency seen
on radiographs has fluid signal on T2-weighted MR ima ges.7,44,45,46,49
on radiographs has fluid signal on T2-weighted MR ima ges.7,44,45,46,49
 FIGURE 25.11 An 8-month-old boy with sickle cell disease dactylitis. The oblique radiograph of foot shows marked periosteal reaction (arrows) and adjacent soft tissue swelling involving the first and second metatarsals. |
The treatment of a vasoocclusive crisis and infarcts from SCD requires the use of analgesics, vasodilators, and aggressive intravenous hydration. If a coexisting infection is suspected, antibiotics are also given.45,50
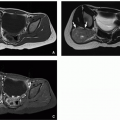
 FIGURE 25.12 A 15-year-old girl with sickle cell disease and multiple painful crisis in the past. A: Coronal T1-weighted MR image of the pelvis shows bilateral proximal femoral serpiginous lesions with fatty elements and a sclerotic low-signal rim indicative of old bone infarcts (asterisks). There is a decreased T1 signal in both proximal femoral diaphyses indicative of bone marrow reconversion (arrows). B: Frontal radiograph of the pelvis demonstrates patchy increased sclerosis (asterisks) in both iliac wings and femoral heads indicating multiple previous bone infarcts. The femoral heads are symmetric in size and shape with preserved sphericity. |
Storage Diseases
Storage diseases are progressive multisystemic metabolic conditions that affect the musculoskeletal system and viscera, such as the lungs, heart, liver, spleen, and in some cases the brain as well. They can be mainly divided into lysosomal and nonlysosomal diseases, with the metabolic substrate determining the involvement of the cell type or organ in the individual storage disease.51 Individually, lysosomal storage diseases (LSDs) are rare genetic conditions; however, the cumulative incidence of lysosomal diseases is 1:5,000, with more than 50 types described. Most LSDs are inherited as an autosomal recessive pattern.52 Disorders of the lysosomal type require electron microscopy of the affected tissue for morphologic diagnosis.51 Lysosomes contain enzymes that degrade cellular macromolecules, and when a specific enzyme is absent, excessive by-products and metabolites accumulate. Typically, LSDs are classified based on the accumulating substrate: mucopolysaccharidoses (MPS), mucolipidoses, sphingolipidoses, glycoprotein storage diseases, and glycogenosis. LSDs are differentiated by clinical features, the age of presentation, and enzyme deficiency. All LSDs affect the skeletal system with overlapping phenotypes; however, some have unique skeletal involvement.52
As with any other skeletal dysplasia, a skeletal survey is the first imaging approach, allowing for the evaluation of the entire skeleton. In the MPS, the accumulation of glycosaminoglycans (GAGs) occurs because of a deficiency in different GAG-degrading enzymes, thus causing progressive damage of affected tissues. Seven distinct clinical types of MPS have been identified, caused by 11 different enzymatic deficiencies (Table 25.1).8,53,54 Mucolipidoses (ML) constitute a more heterogeneous group, with types I and IV being very different from types II and III. Although these conditions are usually grouped together, the enzyme abnormality, stored
substrate, and clinical features are very different. ML II (I-cell disease) (Fig. 25.13) and ML III (pseudo-Hurler polydystrophy) represent two ends of the same clinical spectrum caused by mutations in the gene encoding for the enzyme UDP-N-acetylglucosamine, the latter being the attenuated form with a late presentation. ML I and ML IV are caused by deficiency of alpha-N-acetyl-neuraminidase (sialidase) and mucolipin/alpha-N-acetyl-neuraminidase (sialidase), respectively.55
substrate, and clinical features are very different. ML II (I-cell disease) (Fig. 25.13) and ML III (pseudo-Hurler polydystrophy) represent two ends of the same clinical spectrum caused by mutations in the gene encoding for the enzyme UDP-N-acetylglucosamine, the latter being the attenuated form with a late presentation. ML I and ML IV are caused by deficiency of alpha-N-acetyl-neuraminidase (sialidase) and mucolipin/alpha-N-acetyl-neuraminidase (sialidase), respectively.55
TABLE 25.1 Enzyme Deficiency in Mucopolysaccharidoses and Mucolipidoses | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
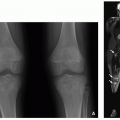
Dysostosis multiplex is a term used to designate the common radiographic manifestations of MPS and ML.8,52,53,55 General skeletal manifestations of MPS, including short stature, joint stiffness, and contractures, are seen in all types of MPS except for Morquio syndrome, where hypermobility is common. In the axial skeleton, there is involvement of the skull, thorax, spine, and pelvis. In the skull, thickening of the vault, macrocephaly with dolichocephaly, and a “J”-shape sella are characteristic. In the face, lack of pneumatization of the paranasal sinuses, a short and broad mandible, and unerupted and widely spaced teeth can be found. In the thorax, characteristic findings include oar-shaped ribs because of anterior rib widening (Schematic H), small scapulae, a short sternum, and thickened clavicles. Spine involvement is common and includes odontoid hypoplasia, atlantoaxial instability, and spinal stenosis, which are more critical in patients with Morquio syndrome (MPS-type IV), in whom spinal cord compression and cervical myelopathy may exist (Fig. 25.14 and Schematic G). The vertebral bodies are oval (bullet shape) and flat (platyspondyly) with anteroinferior beaking in Hurler and Hunter syndromes and central beaking in Morquio syndrome (Fig. 25.14 and Schematic E).
 FIGURE 25.13 Mucolipidosis type II (I-cell disease) in a 3-year-old boy. The vertebral bodies are misshapen and contain multiple Schmorl nodules (left). Microscopically, the foamy storage cells are particularly prominent lining the periosteum (right; hematoxylin and eosin, original magnification, 600×). |
The evaluation of the spinal involvement by MPS is best done using CT scan with three-dimensional (3D) reconstructions to assess spinal morphology and craniocervical junction and with MRI to evaluate the spinal cord in cases of suspected cord compression and myelopathy. These spinal cord abnormalities are best seen on sagittal T1- and T2-weighted MR images. In the pelvis rounded iliac wings with an inferior tapering of the ilium and underdevelopment of the acetabula and femoral heads are seen (Fig. 25.15 and Schematic J). In the peripheral skeleton, the long bones are
notoriously affected with shortening of the diaphysis, hypoplastic epiphyses, irregular and widened physes, proximal humeral notching, and genu valgum due to hypoplasia of the lateral tibial plateaus (Fig. 25.15 and Schematic K). All forms of MPS affect the hands and feet. The metacarpal and metatarsal bones are short and squad with a pointy appearance proximally. The distal radius and ulna are underdeveloped and have a “V”-shape appearance, causing wedging of the carpal bones. Thickening of the subcutaneous soft tissues may result in claw hand deformity due to incomplete extension.8,53,56,57,58
notoriously affected with shortening of the diaphysis, hypoplastic epiphyses, irregular and widened physes, proximal humeral notching, and genu valgum due to hypoplasia of the lateral tibial plateaus (Fig. 25.15 and Schematic K). All forms of MPS affect the hands and feet. The metacarpal and metatarsal bones are short and squad with a pointy appearance proximally. The distal radius and ulna are underdeveloped and have a “V”-shape appearance, causing wedging of the carpal bones. Thickening of the subcutaneous soft tissues may result in claw hand deformity due to incomplete extension.8,53,56,57,58
 FIGURE 25.14 An 8-year-old girl with Morquio syndrome and lower extremity weakness. Sagittal T2-weighted MR image of the spine shows bullet-shaped vertebral bodies with anterior beaking (asterisks). There is marked narrowing (arrows) of the cervical canal and craniocervical junction with increased T2 signal of the cord at that level indicating mild myelopathy. |
Gaucher disease (GD) is an example of glycosphingolipidoses. GD is the most prevalent inherited LSD.59,60 Even though it is most common in Ashkenazi Jews, it can affect individuals from any racial background. It results from a deficient level of the lysosomal enzyme beta-glucocerebrosidase with accumulation of cerebrosides in monocytes and macrophages, called Gaucher cells.59,60,61 Affected pediatric patients frequently complain of bone pain and present with fractures and progressive joint collapse that impair mobility. Osteonecrosis is the most disabling skeletal manifestation of the disease.52,58,59,61
Radiographs are readily available and are inexpensive, but they can be insensitive to the marrow changes in GD. Radiographs are helpful in evaluating fractures and osseous deformities, but MRI is the method of choice for evaluating bone marrow involvement and epiphyseal osteonecrosis. The skeletal manifestations of GD are secondary to the infiltration of the bone marrow with Gaucher cells (Fig. 25.16) and include growth retardation; osteopenia that predispose to stress fractures; classic bone deformity called Erlenmeyer flask deformity, which is seen as flaring of the metaphysis of the long bones; osteonecrosis; and bone infarcts. Areas of osteosclerosis can be seen in old healed bone infarcts. Marrow infiltration of Gaucher cells causes a decreased T2 signal in the otherwise normal yellow marrow. Pyogenic osteomyelitis, although it is uncommon in Gaucher disease, can be difficult to differentiate from bone infarcts.61
 FIGURE 25.15 Mucolipidosis type II (I-cell disease) in a 3-year-old boy. Frontal radiograph of the lower extremities shows classic radiographic findings of mucopolysaccharidosis, which include rounded iliac wings (asterisks) with inferior iliac tapering, underdeveloped acetabula and femoral heads (arrows at the level of hip joint), irregular and widened distal femoral physes (arrows at the level of knee joint), and bilateral genu valgum. |
Optimal management of these multisystemic disorders requires the involvement of a multidisciplinary/multispecialty team, including the radiologist. Enzyme replacement therapy is available for MPS I, II, and VI and is in development for types IV and VII. Enzyme therapy is also available for GD. Stem cell transplantation can preserve cognition and prolong survival in very young children with the more severe types, such as MPS I.54,60
 FIGURE 25.16 Gaucher disease diagnosed via femur and tibia biopsies in an 8-year-old girl who carried a diagnosis of “osteomyelitis” status post multiple debridements. The storage cells expand the marrow space, compressing blood vessels (left) causing osteonecrosis in a manner similar to that seen in osteomyelitis. In this chronic lesion, infarcted bone (“sequestrum”) is encased by viable new bone (right). Hematoxylin and eosin, original magnification, 600× (left) and 200× (right). |
Bone Age Evaluation
Skeletal maturation is marked by an orderly and generally reproducible sequence of recognizable changes in the appearance of the skeleton during childhood. Common indications for evaluating skeletal maturity include suspicion of underlying endocrine disorders, congenital/genetic syndromes, and constitutional short stature and before planning orthopedic procedures, such as scoliosis repair or leg length discrepancy.62,63
Radiographic assessment of skeletal maturity is most commonly based on the appearance of the hand and wrist using an anterior-posterior radiograph of the left hand and wrist. In children who are younger than 2 years of age, the use of knee and ankle radiographs are helpful, as the carpal bones are usually not ossified yet, and there is little change in the appearance of the hand and wrist.62 Skeletal maturity can be estimated by comparing the radiographic appearance of the child’s hand and wrist bones with the radiographic appearance of these bones in a healthy group of children of the same age. The most commonly used methods, both based on the recognition of maturity indicators, are the Greulich and Pyle (G&P) atlas and the Tanner-Whitehouse 2 (TW2) methods.62,63,64 Maturity indicators are based on the initial onset of ossification of the growth centers, the size and positional relationships of the epiphyses and metaphyses, specific modeling changes that affect the epiphyses and metaphyses, and the closure of the growth plates (Fig. 25.17).62,63
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


