Fig. 9.1
Etiologies of valvular stenosis: (a) Calcific aortic valve stenosis. (b) Congenital bicuspid aortic valve stenosis. (c) Rheumatic aortic valve stenosis (Images courtesy of Michael A. Seidman, M.D., Ph.D., Cardiovascular Pathology Fellow, Department of Pathology, Brigham & Women’s Hospital and Harvard Medical School, Boston, MA)
Historical Perspective
The pathologist Lazare Rivière first described aortic valve stenosis in 1663 [2]. A few years later in 1672, Carolus Rayger reported a case of sudden death due to AS [3]. Pathological evaluations in both of these cases demonstrated heavy calcifications of the aortic valve, but the young age of these patients caused physicians to relate their stenotic valves to congenital causes. It was not until 1812 that Napoleon’s physician Jean Corvisart described the clinical manifestations of AS [4] with William Stokes describing the characteristic murmur decades later in 1854 [5]. Following the introduction of cardiac catheterization in 1955, Schnabel characterized the hemodynamic basis of assessing the severity of AS by simultaneous measurement of aortic and left ventricular pressures [6]. Using these pressure measurements, Gorlin developed a formula to calculate the orifice area of the stenotic aortic valve [7], and this became the gold standard technique to quantitate the degree of AS until the development of accurate noninvasive echocardiographic methods.
The early surgical treatments for AS were fraught with high rates of morbidity and mortality. In 1913, Tuffier performed the first operation for AS, a digital commissurotomy, which was of limited success. The era of aortic valve replacement (AVR) started with the implantation of the first prosthetic aortic valve in 1952 by Hufnagel. Introduction of cardiopulmonary bypass in the mid-1950s helped transform AVR into the standard of care for severe AS where the volume has grown to more than 300,000 surgical valve replacements worldwide each year [8]. Transcatheter aortic valve replacement (TAVR) was approved in Europe in 2007, and it is estimated that 60,000 have since been placed worldwide. The Edwards Sapien valve was approved in the USA in 2011 and significant growth in the use of TAVR is anticipated.
Epidemiology
The prevalence of AS steadily increased in the nineteenth century as a result of the rise in the incidence of rheumatic heart disease. Recognition of the association of rheumatic fever with cardiac disease is largely attributable to J. B. Boullaud in 1835. At its peak in 1,900, an estimated 1 in 1,000 people in the USA had the disease, but the incidence subsequently declined dramatically to less than 3,000 cases each year and a mortality of <1 % [9].
The current prevalence of valvular heart disease is not well defined. The difficulty of determining the true prevalence of valvular heart disease stems from the long asymptomatic latent phase of the disease, a factor likely contributing to the significant underdiagnosis in the elderly. Additionally, many studies are affected by a selection bias of patients referred for screening echocardiography and, thus, likely do not capture the true prevalence. Pooled analysis from three large epidemiologic studies provides the best estimates, reporting a 2.5 % prevalence of valvular heart disease in the general population (Table 9.1) [10]. The prevalence of valvular heart disease increases with age. In those above the age of 75 years, the prevalence of valvular heart disease rises to nearly 12 % (Fig. 9.2).
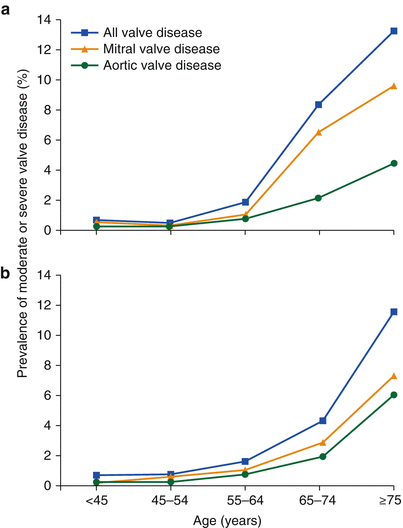
Table 9.1
Prevalence of valvular heart diseases in population-based studies
Age (years) | p value for trend | Frequency adjusted to 2000 US adult population | |||||
|---|---|---|---|---|---|---|---|
18–44 | 45–54 | 55–64 | 65–74 | ≥75 | |||
Participants (n) | 4,351 | 696 | 1,240 | 3,879 | 1,745 | 202,128,094 | |
Male, n (%) | 1,959 (45 %) | 258 (37 %) | 415 (33 %) | 1,586 (41 %) | 826 (47 %) | 100,994,367 (48 %) | |
Mitral regurgitation (n = 449) | 23, 0.5 % (0.3–0.8) | 1, 0.1 % (0–0.8) | 12, 1.0 %(0.5–1.8) | 250, 6.4 % (5.7–7.3) | 163, 9.3 % (8.1–10.9) | <0.0001 | 1.7 % (1.5–1.9) |
Mitral stenosis (n = 15) | 0, 0 % (0–0.1) | 1, 0.1 % (0–0.8) | 3, 0.2 % (0.1–0.7) | 7, 0.2 % (0.1–0.4) | 4, 0.2 % (0.1–0.6) | 0.006 | 0.1 % (0.02–0.2) |
Aortic regurgitation (n = 90) | 10, 0.2 % (0.1–0.4) | 1, 0.1 % (0–0.8) | 8, 0.7 % (0.3–1.3) | 37, 1.0 % (0.7–1.3) | 34, 2.0 % (1.4–2.7) | <0.0001 | 0.5 % (0.3–0.6) |
Aortic stenosis (n = 102) | 1, 0.02 % (0–0.1) | 1, 0.1 % (0–0.8) | 2, 0.2 % (0.6–1.9) | 50, 1.3 % (1.0–1.7) | 48, 2.8 % (2.1–3.7) | <0.0001 | 0.4 % (0.3–0.5) |
Any valve disease | |||||||
Overall (n = 615) | 31, 0.7 % (0.5–1.0) | 3, 0.4 % (0.1–1.3) | 23, 1.9 %(1.2–2.8) | 328, 8.5 % (7.6–9.4) | 230, 13.2 % (11.7–15.0) | <0.0001 | 2.5 % (2.2–2.7) |
Women (n = 365) | 19, 0.8 % (0.5–1.3) | 1, 0.2 % (0.01–1.3) | 13, 1.6 % (0.9–2.7) | 208, 9.1 % (8.0–10.4) | 115, 12.6 % (10.6–15.0) | <0.0001 | 2.4 % (2.1–2.8) |
Men (n = 259) | 12, 0.6 % (0.3–1.1) | 2, 0.8 % (0.1–2.8) | 10, 2.4 % (1.2–4.4) | 120, 7.6 % (6.3–9.0) | 115, 14.0 % (11.7–16.6) | <0.0001 | 2.5 % (2.1–2.9) |
Fig. 9.2
Prevalence of valvular heart disease by age from (a) pooled population-based studies and (b) an Olmsted County community study (From Nkomo et al. [10])
Currently in the USA, degenerative calcific AS comprises the most common cause of AS, largely the result of the aging population. The Euro Heart Survey in 2003 found that among those with AS, 82 % were due to degenerative disease, 11 % from rheumatic causes, and 5 % from congenital causes (Table 9.2) [11]. In underdeveloped countries, however, rheumatic heart disease continues as the major cause of valvular heart disease.
Table 9.2
Etiology of native left-sided single valve disease
Aortic stenosis n = 1,197 | Aortic regurgitation n = 369 | Mitral stenosis n = 336 | Mitral regurgitation n = 877 | |
|---|---|---|---|---|
Degenerative (%) | 81.9 | 50.3 | 12.5 | 61.3 |
Rheumatic (%) | 11.2 | 15.2 | 85.4 | 14.2 |
Endocarditis (%) | 0.8 | 7.5 | 0.6 | 3.5 |
Inflammatory (%) | 0.1 | 4.1 | 0 | 0.8 |
Congenital (%) | 5.4 | 15.2 | 0.6 | 4.8 |
Ischemic (%) | 0 | 0 | 0 | 7.3 |
Other (%) | 0.6 | 7.7 | 0.9 | 8.1 |
Pathophysiology
The understanding of the pathophysiology of degenerative AS has significantly advanced with improved understanding of the cellular mechanisms and the process of calcification [12, 13]. Leaflet thickening and calcification, which progress to calcified nodules, form the hallmark of calcific AS (Fig. 9.3). These calcium deposits eventually compromise valve leaflet mobility, in effect narrowing the effective valve orifice. The pathological findings are well described and reviewed in a separate section of this text.
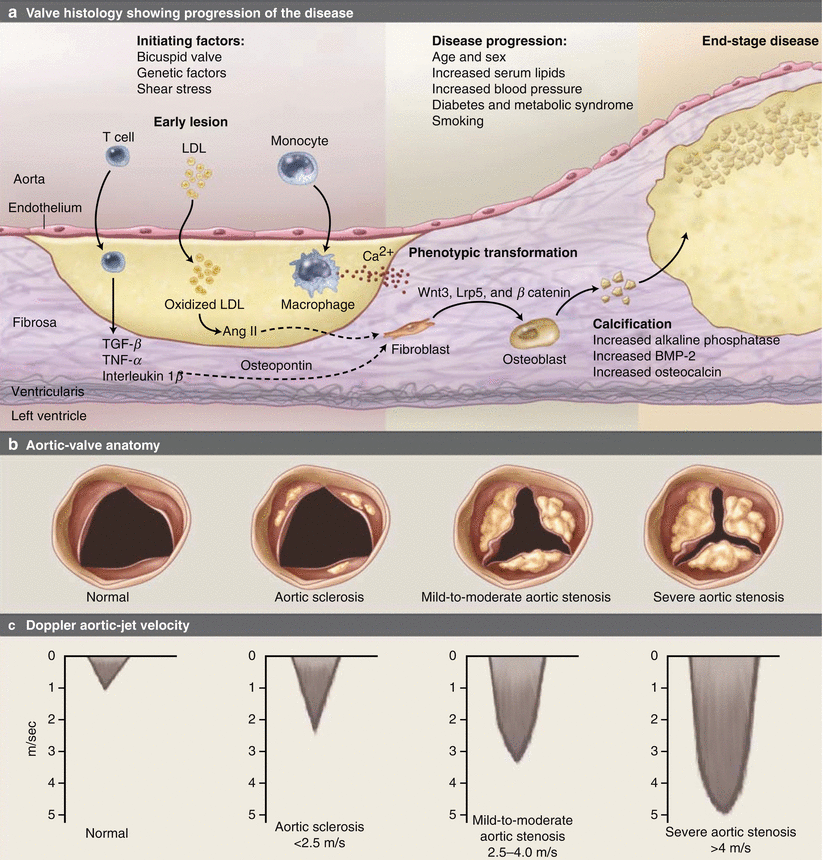
Fig. 9.3
Pathophysiology of calcific aortic stenosis. (a) The histology of the early lesion is characterized by a subendothelial accumulation of oxidized low-density lipoprotein (LDL), production of angiotensin (Ang) II, and inflammation with T lymphocytes and macrophages. Disease progression occurs by several mechanisms including local production of proteins, such as osteopontin, osteocalcin, and bone morphogenetic protein 2 (BMP-2), which mediate tissue calcification; activation of inflammatory signaling pathways including tumor necrosis factor-α (TNF-α), tumor growth factor-β (TGF-β), the complement system, C-reactive protein, and interleukin-1β; and changes in tissue matrix including the accumulation of tenascin C, and upregulation of matrix metalloproteinase-2 and alkaline phosphatase activity. In addition, leaflet fibroblasts undergo phenotypic transformation into osteoblasts, regulated by the Wnt3-Lrp5-β catenin signaling pathway. Microscopic accumulations of extracellular calcification (Ca2+) are present early in the disease process, with progressive calcification as the disease progresses and areas of frank bone formation in end-stage disease. The corresponding changes in the aortic valve anatomy are viewed from the aortic side with the valve open in systole (b) and the corresponding Doppler aortic jet velocity (c) (From Otto [142])
Current evidence suggests that oxidative stress initiates early valve sclerosis. The combination of lipid deposition, neuroendocrine, and hemodynamic factors may propagate these changes. In susceptible patients, valvular interstitial and endothelial cells develop an osteoblastic phenotype and begin producing pro-calcific mediators, collagen, and osteopontin to ultimately promote bone formation. The progression of disease is due to both oxidant stress and inflammation causing an increase in extracellular matrix deposition and calcification. Despite the known role of the renin-angiotensin system and lipids, clinical trials have not demonstrated the effectiveness ACE inhibitors or statins in slowing the progression of AS [14–20]. Recent evidence suggests that genetic factors, particularly inflammatory and lipid markers such as IL-10 and Apo B, may be important predictors of calcific aortic disease [21].
Progression to severe AS occurs gradually over a period of several decades. As obstruction of the left ventricular outflow worsens, the left ventricle (LV) remodels to compensate for the significant systolic pressure overload. This remodeling leads to development of concentric left ventricular hypertrophy where the ventricular chamber remains a normal size despite an increase in the wall thickness [22, 23]. The resulting increase in wall thickness allows LV to maintain normal wall stress (i.e., afterload) in the setting of elevated chamber pressures and a normal ejection fraction [24]. However, a normal ejection fraction does not necessarily indicate normal contractility, as the former may be the augmented by preload reserve. When the ventricular hypertrophy cannot adequately compensate for the degree of pressure overload due to severe outflow obstruction, the ejection fraction begins to decline [24, 25]. At this point, decreased myocardial contractility potentially contributes to the depressed ejection fraction as well [26].
As a consequence of left ventricular hypertrophy and interstitial fibrosis of the cardiac myocytes, the left ventricular chamber compliance decreases causing diastolic dysfunction [27, 28]. The increased stiffness of the LV creates higher pressures at all volumes requiring elevation in diastolic filling pressures. In this setting, a prominent a-wave is seen on the pressure tracing due to its more forceful contraction and the decreased compliance of the LV. Indeed, left atrial contraction contributes a great deal to left ventricular filling in these situations [29]. This becomes important in situations where left atrial contraction is lost and can lead to a significant decline in left ventricular performance. After AVR to relieve the left ventricular outflow obstruction, the hypertrophic LV may regress though the concentric stiffness from residual interstitial fibrosis still results in diastolic dysfunction [27].
In the absence of significant volume overload, patients with AS typically experience the classic symptoms on exertion. The mechanisms for development of systolic and diastolic heart failure are discussed above. Syncope probably occurs as a result of a combination of mechanisms [30]. The most direct mechanism relates to the inability to augment cardiac output adequately in the presence of a fixed outflow obstruction. During exercise, the deficit of cardiac output in the setting of a drop in systemic arterial resistance causes a fall in blood pressure and syncope. Various arrhythmias including atrial flutter, atrial fibrillation, and ventricular tachycardia have been observed in patients with AS during episodes of syncope. Additional postulated mechanisms include a vasodepressor effect from left ventricular baroreceptor activation as a response to highly elevated LV pressures during exercise [31].
A suggested mechanism for angina in AS focuses on the oxygen supply/demand mismatch that is accentuated during exercise or periods of tachycardia to cause subendocardial ischemia. The hypertrophied LV in AS consumes more oxygen due to the increased myocardial muscle mass, increased work from systolic pressure overload, and prolonged ejection period due to LV outflow obstruction. At rest, coronary blood flow, while elevated in absolute terms, may be normal or reduced relative to the myocardial mass. Additionally, the hypertrophied ventricle also has a limited coronary flow reserve even in the absence of significant epicardial coronary disease [32, 33]. The elevated intraventricular pressure increases the transmyocardial pressure, which can compress the coronary microcirculation and affect coronary perfusion, especially when coronary perfusion pressure is exceeded [34]. The prolonged ejection period also limits the length of diastole and coronary perfusion.
Natural History
The etiologies of congenital AS include unicuspid, bicuspid, or tricuspid valve anatomy. Unicuspid valves cause severe stenosis at birth while bicuspid disease presents in middle age to elderly. Despite this earlier presentation of symptomatic disease, up to 20 % of elderly patients 80 years and older with AS have bicuspid aortic valves [35]. Bicuspid aortic valves are commonly associated with dilation of the ascending aorta that can lead to aneurysm formation requiring surgical repair. Additionally, bicuspid aortic valves may cluster within families, exhibiting an autosomal dominant with incomplete penetrance pattern of heredity [36]. Tricuspid congenital aortic valve disease is less common and difficult to distinguish from acquired disease. Of the acquired causes, rheumatic disease typically presents in the fifth and sixth decade, with younger presentation in underdeveloped countries [37]. Degenerative calcific aortic valve disease is a disease of the elderly increasing significantly with age above 65 years.
Patients with AS progress through the various stages of disease over a period of many decades. The spectrum of disease includes aortic sclerosis (thickened and calcified valve leaflets without obstruction) at the mild end and moves through worsening degrees of obstruction to severe AS at its most severe. Progression of disease is highlighted by a long latent phase lasting decades during which patients remain asymptomatic and have a low risk of cardiac or sudden death [38, 39]. However, the rate of survival rapidly declines following the onset of symptoms (Fig. 9.4).
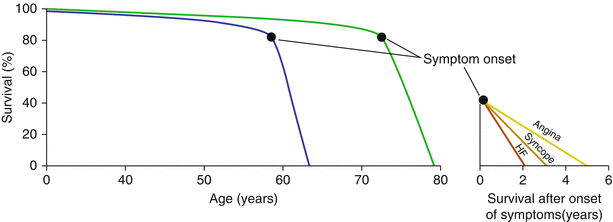
Fig. 9.4
Natural history of aortic stenosis. The blue line represents the disease course as described by Ross and Braunwald. In contemporary times, the disease course may more closely represent the green line. The average survival after development of heart failure (HF), syncope, or angina is shown in the inset (Figure derived from Ross et al. [38])
An estimated 26 % of the population over the age of 65 years old have aortic sclerosis [40]. Histologic data suggests that aortic sclerosis occurs through a degenerative process similar to that of calcific AS [41–45]. Despite the absence of a significant hemodynamic effect on cardiac performance, aortic sclerosis is not an entirely benign lesion. Patients with aortic sclerosis identified on echocardiogram have a 50 % increase in the risk of myocardial infarction and death from cardiovascular causes [46]. A significant portion of patients with aortic sclerosis eventually develop AS. Up to a third of patients with aortic sclerosis eventually progress to having at least mild AS and that 2.5 % of patients with aortic sclerosis ultimately develop severe AS [47–49]. Although the time to progressing from aortic sclerosis to severe AS varies widely, the majority of patients progressing to severe AS do so around 8 years after diagnosis of aortic sclerosis [47].
Once AS is present, the severity of the valve stenosis invariably worsens with time. In patients with moderate AS, the valve area decreases an average of 0.1 cm2 per year, corresponding to a 0.3 m/s increase in jet velocity and a 7 mmHg increase in the mean gradient [50]. Some studies also suggest that AS worsens more rapidly in those with degenerative calcific AS compared with those having congenital or rheumatic aortic valve stenosis [51].
At the onset of symptoms, survival of patients with AS decreases precipitously. Braunwald and Ross reported that in patients with severe AS, death occurs approximately 2 years after the onset of heart failure, 3 years after syncope related to valve stenosis, and 5 years after developing angina (Fig. 9.4) [38]. Subsequent studies have consistently demonstrated similarly poor prognosis of patients with symptomatic severe AS who do not undergo AVR [39, 52–61]. In the PARTNER trial, death occurred in nearly 70 % of patients randomized to contemporary medical therapy over 2 years, which included the use of balloon aortic valvuloplasty [60, 61].
Hemodynamic Assessment
When the physical examination raises suspicion for AS, the next step of the evaluation involves hemodynamic measurements to determine the severity of the valve stenosis. Historically, cardiac catheterization represented the diagnostic standard in hemodynamic assessment because of its ability to obtain direct, real-time pressure measurements of both the LV and aorta (Fig. 9.5). Obtaining a mean transvalvular gradient and cardiac output allows for calculation of the aortic valve orifice area [7]. Noninvasive hemodynamic assessment by echocardiography has largely replaced invasive catheterization and offers several potential advantages [62]. The transaortic valve gradient can be determined by Doppler data and the aortic valve orifice area determined through the concept of flow conservation via the continuity equation. Table 9.3 details the criteria for classifying the severity of AS [62]. Both measures correlate well to catheterization data and have been validated in multiple studies. Echocardiography also allows for direct visualization of the aortic valve anatomy, assessment of LV function, and evaluation for coexisting valvular lesions. Certain situations such as severe hypertension, pressure recovery in small aortas, and low-flow, low-gradient states may affect the accuracy of the echocardiographic assessment of AS. In these cases and in those where noninvasive studies either do not provide adequate assessment or do not correlate with clinical findings, hemodynamics by cardiac catheterization may be helpful in characterizing the severity of the valve stenosis.
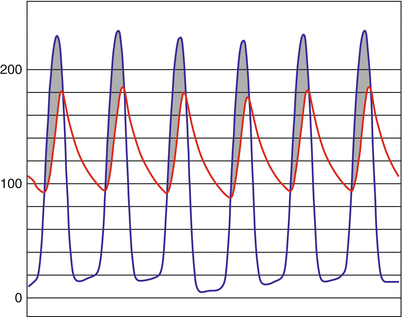
Fig. 9.5
Simultaneous left ventricular (blue) and aortic (red) pressures as measured by cardiac catheterization. The shaded region (gray) highlights the transaortic valve gradient
Table 9.3
Classification of aortic stenosis severity
Mild | Moderate | Severe | |
|---|---|---|---|
Aortic jet velocity (m/s) | 2.6–2.9 | 3.0–4.0 | >4.0 |
Mean gradient (mmHg) | <20a (<30) | 20–40a (30–50b) | >40a (>50b) |
AVA (cm2) | >1.5 | 1.0–1.5 | <1.0 |
Indexed AVA (cm2/m2) | >0.85 | 0.6–0.85 | <0.6 |
Velocity ratio | >0.50 | 0.25–0.50 | <0.25 |
Asymptomatic Patients
The gradual progression of the severity of outflow tract obstruction occurs over a long period, perhaps 10–15 years. During this latent period, clinical outcomes of patients with mild to moderate stenosis remain favorable [63]. The prognosis of patients with moderate to severe stenosis also remains favorable as long as they remain asymptomatic [63]. The risk of sudden death in asymptomatic patients is less than 1 % per year [64]. However, close follow-up is mandatory, as the disease will inevitably progress. The degree of stenosis at which patients develop symptoms is variable with symptomatic patients generally having more severe stenosis [65]. The rate of progression to symptomatic AS has been studied extensively. The strongest predictor of symptom development is the Doppler velocity of the antegrade aortic valve jet [64, 66, 67]. Those with moderate to severe aortic valve calcification or a rapidly increasing aortic valve velocity also have poor clinical outcomes [63]. Furthermore, in asymptomatic patients with very severe AS (peak antegrade aortic jet velocity of ≥5.0 m/s by echocardiography), AVR or cardiac death occurred in 36 % at 1 year and 64 % at 2 years [59]. In that study, the subgroup of patients with a Doppler velocity of ≥ 5.5 m/s had the worst outcomes, with the rate of AVR or death occurring in 56 % at 1 year and 75 % at 2 years.
Patients with asymptomatic AS may underestimate the degree of their symptoms, making evaluation and management difficult for the physician. As symptoms develop over a long period of time, patients may slowly self-limit and not perceive the development of symptoms or simply attribute progressive deconditioning to natural aging. In evaluating patients with asymptomatic severe AS, exercise stress testing and stress echocardiography may help with assessment of exercise capacity and functional valve hemodynamics to guide decision-making [68]. Risk scores may also help with assessment of asymptomatic patients. Female sex, peak aortic velocity, and B-type natriuretic peptide independently predict need for AVR or death within 24 months, suggesting that valve interventions could also benefit select asymptomatic patients with severe AS [69].
Symptomatic Patients
When AS becomes moderate to severe, patients may present with the classic symptoms of angina, exertional dyspnea or congestive heart failure, and syncope. At this stage, patients have a poor long-term prognosis if the LV outflow obstruction is not treated. In addition to angina due to the effects of ventricular hypertrophy from AS, patients often have angina from concomitant coronary atherosclerosis. Up to half of patients with severe AS also have significant coronary artery disease [70, 71]. Accordingly the ACC/AHA valvular heart disease guidelines suggest coronary angiography in patients over the age of 35 with risk factors for coronary disease being considered for AVR (Box 9.1) [72]. While patients commonly experience dyspnea with exertion, frank congestive heart failure typically occurs during later stages of the disease when ventricular function deteriorates and cardiac output declines from chronic LV outflow obstruction. In situations where poor LV function and decreased cardiac output result in low aortic valve gradients—termed low-flow, low-gradient AS—estimating the degree of AS becomes challenging. The characteristic late peaking systolic murmur of aortic valve stenosis becomes subtle and easy to overlook. In the elderly patients, other typical physical findings such as parvis and tardis pulse, single second heart sound, and a precordial thrill may be absent. On echocardiography, motion of a calcified aortic valve may appear restricted due to the low cardiac output of a poorly functioning ventricle alone. Using dobutamine to augment cardiac output aids in differentiating this “pseudo AS” from true severe AS [68]. Patients displaying contractile reserve with dobutamine also have more favorable outcomes, including improvement in LV function, after AVR [73–76]. Invasive hemodynamic assessment by cardiac catheterization can also help in assessment in these patients.
Box 9.1 ACC/AHA Guideline Indications for Aortic Valve Replacement [72]
Class I | AVR is indicated for symptomatic patients with severe AS. (Level of Evidence: B) |
AVR is indicated for symptomatic patients with severe AS undergoing coronary artery bypass graft surgery. (Level of Evidence: C) | |
AVR is indicated for symptomatic patients with severe AS undergoing surgery on the aorta or other heart valves. (Level of Evidence: C) | |
AVR is recommended for patients with severe AS and LV systolic dysfunction (ejection fraction less than 0.50). (Level of Evidence: C) | |
Class IIa | AVR is reasonable for patients with moderate AS undergoing CABG or surgery on the aorta or other heart valves. (Level of Evidence: B) |
Class IIb | AVR may be considered for asymptomatic patients with severe AS and abnormal response to exercise (e.g., development of symptoms or asymptomatic hypotension). (Level of Evidence: C) |
AVR may be considered for adults with severe asymptomatic if there is a high likelihood of rapid progression (age, calcification, and CAD) or if surgery might be delayed at the time of symptom onset. (Level of Evidence: C) | |
AVR may be considered in patients undergoing CABG who have mild AS when there is evidence, such as moderate to severe valve calcification, that progression may be rapid. AVR may be considered for asymptomatic patients with severe AS and abnormal response to exercise (e.g., development of symptoms or asymptomatic hypotension). (Level of Evidence: C) | |
AVR may be considered for asymptomatic patients with extremely severe AS (aortic valve area less than 0.6 cm2, mean gradient greater than 60 mmHg, and jet velocity greater than 5.0 m/s) when the patient’s expected operative mortality is 1.0 % or less. AVR may be considered for asymptomatic patients with severe AS and abnormal response to exercise (e.g., development of symptoms or asymptomatic hypotension). (Level of Evidence: C) | |
Class III | AVR is not useful for the prevention of sudden death in asymptomatic patients with AS who have none of the findings listed under the Class IIa/IIb recommendations. (Level of Evidence: B) |
Medical Treatment
Medical treatment of symptomatic AS offers limited benefit as no current therapy has demonstrated the ability to alter the disease course. The contemporary understanding of calcific AS attributes valve calcification to an active process rather than a passive degenerative one. Several studies have demonstrated the association of calcific AS with the traditional risk factors and cellular mechanisms of atherosclerosis [15, 40, 43, 45, 77–81]. Despite the suggestion that these risk factors may represent targets of therapy [82, 83], none have yet effectively delayed or prevented progression of aortic valve stenosis. While reports had showed the potential benefit of statins and angiotensin-converting enzyme inhibitors in preventing disease progression [14–19], the large-scale, prospective, randomized trial failed to demonstrate a benefit of using atorvastatin to slow the progression of AS over 3 years of follow-up [20].
Related posts:
 Transcatheter Aortic Valve Replacement: Current Evidence from Large Multicenter Registries
Transcatheter Aortic Valve Replacement: Current Evidence from Large Multicenter Registries
 Positron Emission Tomography Evaluation of Aortic Stenosis
Positron Emission Tomography Evaluation of Aortic Stenosis
 MRI Evaluation of Aortic Stenosis
MRI Evaluation of Aortic Stenosis
 Fusion Imaging for TAVR
Fusion Imaging for TAVR
 Aortoiliofemoral Assessment: MDCT
Aortoiliofemoral Assessment: MDCT
 Valve-in-Valve for Transcatheter Aortic Valve Replacement: Do Imaging Requirements Change?
Valve-in-Valve for Transcatheter Aortic Valve Replacement: Do Imaging Requirements Change?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


