Neuroblastoma (NB) is a malignancy derived from embryonic neural crest cells of the peripheral sympathetic nervous system. It is the most common extracranial solid tumor of childhood, accounting for 15% of cancer-related deaths. The behavior of NB is marked by clinical heterogeneity, which leads to differences in behavior ranging from spontaneous maturation in some patients to inexorable rapid metastatic progression in others. It was noted as early as 1927 to be a tumor in which spontaneous maturation could occur, when Cushing and Wolbach (1) reported the case of a 2-year-old boy whose thoracic paravertebral sympathetic NB, over the course of 10 years, transformed into a completely differentiated ganglioneuroma. However, advanced NB is the most lethal of childhood solid tumors, often resistant to all attempts at disease eradication (2). The enigmatic behavior of this tumor is beginning to be elucidated by new research into the genetic and biologic diversity, but the treatment of advanced disease is a continuing challenge. This chapter discusses the biologic features of NB associated with differing behavior, the evaluation and staging, and a risk-based approach to therapy, with an emphasis on local control issues.
EPIDEMIOLOGY AND SCREENING
There are approximately 650 cases of NB annually in the United States, with an overall incidence of 10.3 per million per year from birth to the age of 15 years. NB accounts for nearly 10% of pediatric cancer in children younger than 15 years, making it the fourth most common malignancy in children after leukemias, brain tumors, and lymphomas. However, it is the most common malignancy of children younger than 18 months, with an incidence of 29.1 per million per year in children younger than 5 years (3). Ninety percent of patients with NB are diagnosed by 10 years, 79% before 4 years, and 36% are infants less than 1 year. The incidence is slightly higher in boys than in girls. The median age at diagnosis is 22 months. NB is responsible for 15% of childhood cancer mortality, with an annual mortality rate of 5 per million for all children and 9 per million for the 0- to 4-year-old subgroup (3).
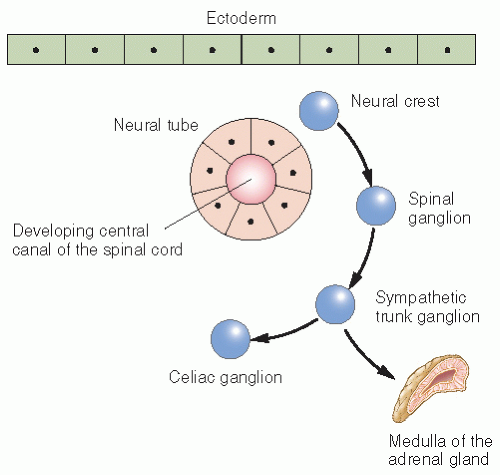
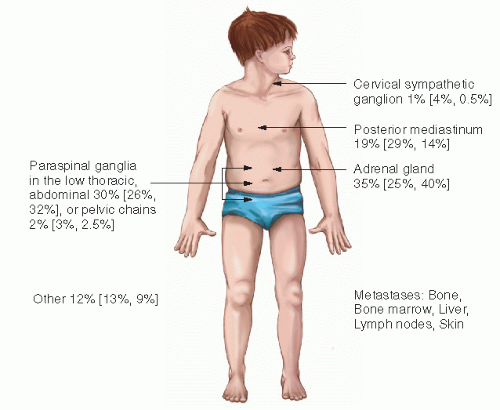
NB arises from primitive (fetal) adrenergic neuroblasts of neural crest tissue, which may explain its high incidence in infancy. In the embryo, continuous columns of neural crest tissue form dorsolateral to the developing neural tissue. These columns are the precursors of the spinal ganglia, the dorsal spinal nerve roots, and the chromaffin cells, which flank the abdominal aorta (4). The largest of these masses is the adrenal medulla (Fig. 6.1). Most cases of NB occur in an anatomic distribution consistent with the location of neural crest tissue (Fig. 6.2).
The frequency of neuroblastic nodules, resembling NB in situ, in the adrenals of autopsied infants who have died of other causes ranges from 1 in 39 to 1 in 600 autopsies, suggesting that most of these nodules regress by birth (5). This is interpreted to mean that such nodules are embryologic adrenal remnants and are the cells from which adrenal medulla NB may develop. Several studies using systematic screening of neonates for NB have confirmed the results of earlier autopsy studies by suggesting a much higher incidence of NB than is clinically evident. More than 90% of NBs secrete vanillylmandelic acid (VMA) or homovanillic acid (HVA) in the urine, for which rapid quantitative screening methods have been developed that have a high degree of sensitivity and specificity. Urinary catecholamine screening for NB was initially proposed by Woods et al. (6) because the incidence of 8.7 per 1 million children per year is comparable to or higher than that of other congenital diseases for which screening is already in place, such as hypothyroidism, galactosemia, and phenylketonuria. The poor prognosis for advanced NB in children older than 12 months at diagnosis compared with the favorable outcome for those diagnosed in infancy raised the expectation that diagnosis at an earlier age might improve overall outcome. However, the cumulative data from 30 years of screening in Japan, the Quebec Neuroblastoma Screening Project, and screening projects in Europe have shown that screening infants by 6 months of age results in a significant increase in the overall incidence of NB but fails to reduce the incidence of advanced-stage disease with poor prognosis (7, 8, 9). NBs detected by screening have almost exclusively favorable biologic features (MYCN nonamplified, triploid, favorable Shimada histology) (10,11). Thus, screening practices result in the overdiagnosis of tumors that would otherwise have spontaneously regressed. A recent publication from the Quebec project conclusively showed that screening at 3 weeks and 6 months in a cohort of 476,654 infants born over a 5-year period had no impact on overall mortality for the screened cohort compared with multiple control populations from Minnesota, the Delaware Valley, Florida, and the rest of Canada, verifying the results of the earlier studies (7). A second study screened 1,475,773 children at 1 year of age in German states and similarly revealed no impact on outcome (12). The screened group and children in the control area had a similar incidence of stage 4 NB (3.7 cases per 100,000 screened children and 3.8 per 100,000 controls) and a similar rate of death among children with NB (1.3 deaths per 100,000 screened children and 1.2 per 100,000 control subjects). There was a substantial rate of overdiagnosis in the screened group of children without benefit from the screening.
Figure 6.1 Migration of the neural crest in the human embryo.
Figure 6.2 Common locations of neuroblastoma. The first percentage is the overall proportion of cases at the site. The numbers in parentheses are the percentages of cases at a given site in children 1 year of age or younger and older than 1 year of age. (Data are from Bernstein ML, Leclerc JM, Bunin G, et al. A population-based study of neuroblastoma incidence, survival, and mortality in North America. J Clin Oncol. 1992;10(2):323-329; and J. Shuster, PhD, Pediatric Oncology Group Statistical Office.)
The environmental and inherited factors involved in the pathogenesis of NB are largely unknown, despite extensive epidemiologic and genetic studies. Given the young age at which most NBs present, it has been suggested that environmental exposures before conception or during pregnancy increase the risk of NB. Epidemiologic investigations have implicated fetal exposure to diuretics, tranquilizers, hormones, phenytoin, alcohol, and tobacco as increasing the risk of NB. However, these studies have lacked the statistical power to convincingly demonstrate that these drugs are etiologic risk factors or have not been confirmed by subsequent studies (13). Environmental exposures are unlikely to be significant, although certain parental occupations have been associated with increased risk of NB, including those involving electrical exposures, gardening, farming, and painting (14, 15, 16). However, none of these associations has been seen consistently.
Although NB usually occurs sporadically, 1-2% of patients have a family history of the disease. This is similar to the other embryonal cancers of childhood in which a familial predisposition is observed. Familial NB is inherited in an autosomal dominant Mendelian fashion with incomplete penetrance. Affected children from these families differ from those with sporadic disease in that they are often diagnosed at an earlier age (usually infancy) or they have multiple primary tumors. These clinical characteristics are hallmarks of the two-mutation cancer predisposition model first proposed for retinoblastoma. Therefore, it seems likely that familial NB occurs as a result of a germ-line mutation in one allele of a tumor suppressor gene. Germ-line mutations have rarely been identified, with only rare case reports of familial cases with mutations in 1p36, one of the common areas of allelic loss in sporadic NB (17). Other cases may result from different hereditary-predisposition loci, now attributed to multiple different genetic aberrations. Chromosome 16p12-13 was identified as a likely predisposition locus, although no causal gene has been identified (18). NB has also been seen in several patients with constitutional chromosomal rearrangements, including deletions overlapping putative tumor suppressor loci at chromosome bands 1p36 and 11q14-23. NB has also been reported in patients with other neural crest disorders such as the Hirschsprung disease and central hypoventilation syndrome; these cases are associated with a germ-line mutation at chromosome 4p12 (PHOX2B gene) (19). More recently, multiple groups have identified the anaplastic lymphocyte kinase (ALK) gene as a predisposition gene in familial NB, and also frequently mutated in tumors from sporadic NB (20, 21, 22).
BIOLOGY
Cytogenetic and molecular studies in NB are providing new insights into its biology and prognosis. Unfavorable biologic features include amplification of MYCN, deletion or loss of heterozygosity of chromosome 1p or 11q, and gains at 17q, and favorable biologic features include hyperdiploidy and overexpression of Trk A (23, 24, 25, 26, 27, 28, 29) Recognizing these biologic pathways in NB has improved the ability to treat children with risk-based therapy and may lead to novel therapeutic approaches in the near future.
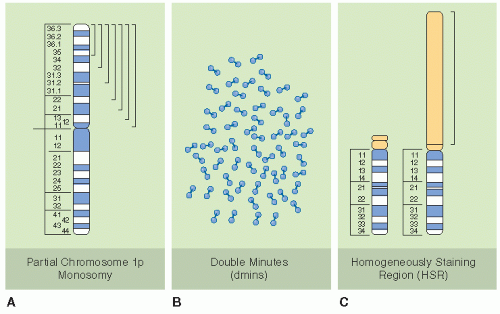
MYCN amplification was the first and is now the most widely accepted biologic marker of prognosis for NB (30,31). MYCN is in the family of MYC oncogenes that encode transcriptional regulatory factors involved in the control of other genes. The amplification is generally a consequence of an aberrant gain of multiple copies of the MYCN gene. Homogeneously staining regions (HSRs, nonbanding regions of metaphase chromosomes that stain homogeneously) and double-minute chromatin bodies (fragments of HSRs) are a manifestation of gene amplification (Fig. 6.3). They are derived from the distal short arms of chromosome 2, which contains the proto-oncogene MYCN. The excess number of MYCN genes leads to overexpression of the n-myc protein and subsequent overproduction of growth-promoting signals and stimulation of proliferation and tumorigenesis (23). Mice with aberrant overexpression of MYCN develop NB, providing evidence that MYCN plays a prominent role in NB development (32). MYCN amplification is most easily detected on fluorescent in situ hybridization of fresh tumor imprints, although it may also be tested by immunohistochemistry, Southern blot, or polymerase chain reaction. Recent studies also show that circulating MYCN DNA can be detected in advanced-stage NB with MYCN amplification with a sensitivity of 75-85% (33).
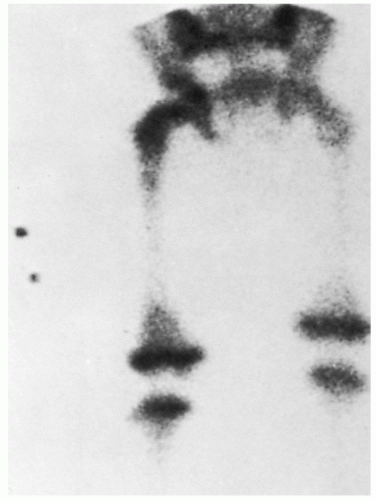
Figure 6.3 Common cytogenetic abnormalities in human neuroblastomas. Shown are diagrammatic representations of the three most common cytogenetic abnormalities seen in human neuroblastomas. A: Deletions of the short arm of chromosome 1. The brackets indicate that the region deleted in different tumors is variable in terms of its proximal breakpoint, but the distal short arm appears to be deleted in all cases, resulting in partial 1p monosomy. B: Extrachromosomal double-minute chromatin bodies (dmins). These are seen in about 30% of primary neuroblastoma and are a cytogenetic manifestation of gene amplification. C: Homogeneously staining region (HSR). A representative HSR on the short arm of chromosome 13 is shown in this example. HSRs are a cytogenetic manifestation of gene amplification in which the amplified sequences are chromosomally integrated. (From Bernstein ML, Leclerc JM, Bunin G, et al. A population-based study of neuroblastoma incidence, survival, and mortality in North America. J Clin Oncol 1992;10(2):323-329; and Powis MR, Imeson JD, Holmes SJ. The effect of complete excision on stage III neuroblastoma: a report of the European Neuroblastoma Study Group. J Pediatr Surg 1996;31(4):516-519, with permission.)
Amplification of MYCN in primary NB has been shown to correlate with advanced stage and a poor prognosis. Such amplification occurs in 30-40% of advanced-stage NB but in only 5-10% of low-stage or 4S disease and not at all in benign ganglioneuromas (34). Children who have MYCN amplification in their tumor need more intensive or novel therapy to control the disease. MYCN usually is an independent prognostic factor such that amplification portends an unfavorable outcome, even in disease settings that would otherwise be favorable. MYCN amplification is seldom identified in localized stage disease (stage 1 and 2) but may predict a poor outcome in these patients when present (35). Amplification of MYCN is also uncommon in infants with stage 4S disease but predicts a less favorable survival (44% vs. 90%) (36). Amplified MYCN is more common in tumors from children older than 1 year with advanced stage and elevated ferritin, neuron-specific enolase, lactate dehydrogenase, and chromosome 1p deletion (34).
DNA ploidy is also an important discriminator of response to chemotherapy for NB. In a 1991 report, Look et al. (27) evaluated 298 children. In infants with metastatic disease, the progression-free survival (PFS) was more than 90% for those with hyperdiploid tumors and 0% in diploid cases. In children 12-24 months of age with stage D, the distinction was also striking (50-60% in hyperdiploid cases and 0% in diploid, p < 0.001). A subsequent Pediatric Oncology Group (POG) study of infants with unresectable or metastatic NB reported an overall 3-year survival of 94% for infants with hyperdiploid tumors and 55% for diploid (37). Hyperdiploid tumors also appear to counteract the unfavorable impact of MYCN amplification in low-stage tumors (28). Ploidy is probably less useful as a prognostic marker in children older than 24 months, in whom most tumors are diploid.
Specific chromosomal deletions often are present in NB. Deletion of the short arm of chromosome 1 occurs in 30-50% of primary tumors and usually lies at the distal end of chromosome 1 in the area of 1p36. Loss of chromosome 1p strongly correlates with MYCN amplification and is also associated with a poor prognosis. Loss of heterozygosity of 1p in tumors without MYCN amplification is an independent prognostic marker of outcome, although this prognostic value is lost in MYCN-amplified tumors (24).
Loss of heterozygosity has also been reported for chromosome 11q and is identified in nearly half of the NB samples analyzed. Unbalanced loss of chromosome 11q is associated with a worse prognosis and is inversely related to MYCN amplification (24). Chromosomal deletions may result in the loss of a suppressor gene. Several candidate genes for the NB suppressor gene have been mapped to 1p36 (38) and 11q; however, none have been shown to have mutations in the nondeleted allele. An alternative pathway for tumorigenesis from one of the candidate genes may relate to dosage effect, with the allelic loss causing a reduction in the level of an important protein.
Partial gains at chromosome 17q are another common genetic alteration and are associated with an adverse outcome. Gains on chromosome 17q have been linked to several prognostic factors: age greater than 1 year, advanced-stage disease, deletion of chromosome 1p, and amplification of MYCN. It is the most common genetic alteration found in NB, occurring in 50% of cases (39). In one study, patients with the gain of 17q had a 5-year relapse-free survival of 16%, compared with 75% in patients without this genetic alteration (25). Again, whether 17q gain is an important independent marker of prognosis must be verified in a larger prospective study.
In addition to gains and losses of genetic material, other genes that have been identified as prognostically important include nerve growth factor receptor expression, telomerase activity, and genes involved in invasion and metastasis. The neurotrophins and their receptors are important in nervous system development and hence NB tumorigenesis. Expression of Trk A and Trk C has been found to correlate with lower stage and the absence of MYCN amplification. Conversely, expression of Trk B correlates with amplification of MYCN. The balance of Trk expression may influence the spontaneous regression of NB or the differentiation into benign ganglioneuromas (40). High levels of telomerase activity may also predict an aggressive tumor phenotype. Integrins are also important in NB. The majority of NB tumors express high levels of CD44, except for tumors of advanced stage and MYCN amplification. Lack of CD44 expression is strongly associated with MYCN amplification and, in multivariate analysis, is an independent predictor of overall survival (OS) (23). Another integrin, alphaVbeta3, appears to be expressed predominantly in undifferentiated and high-risk NB (41), with an antiangiogenic integrin alphaV antagonist able to inhibit growth of NB in a syngeneic murine tumor model by working synergistically with an anti-GD2 antibody (42,43). The metalloproteinases MMP2 (gelatinase A) and MMP9 (gelatinase B) are expressed in primary NB tumors, with increased expression associated with more advanced stage and MMP2 correlating with worse outcome (44). The angiogenic vascular endothelial growth factor has been found in both NB cell lines and primary tumors, with higher expression in high-risk disease (45).
PATHOLOGY
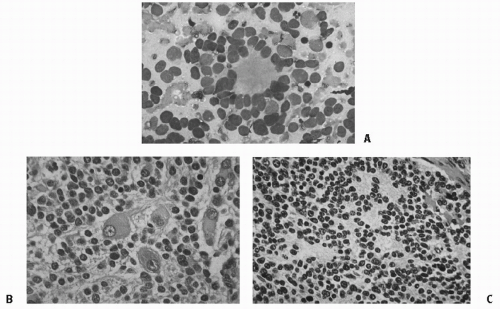
NB is one of the small blue round cell tumors, along with the non-Hodgkin lymphoma, the Ewing sarcoma, undifferentiated soft tissue sarcomas including rhabdomyosarcoma, and primitive neuroectodermal tumors. The classic histologic subtypes of neuroblastic tumors include NB, ganglioneuroblastoma, and ganglioneuroma, reflecting a pattern of increasing maturation and differentiation (46). The cells of NB are small and uniformly sized, with dense hyperchromatic nuclei and scant cytoplasm (Fig. 6.4). The cells may be densely packed, separated by thin fibrils or bundles, and necrosis and calcification can occur. Neuritic processes can be demonstrated in most cases, and pseudorosettes can be seen in 15-50% of cases. The other end of the spectrum, ganglioneuroma, has mature ganglion cells, neuritic processes, and Schwann cells and has more fibrillary material. Patients with ganglioneuroma or ganglioneuroblastoma generally have localized tumors with favorable biologic characteristics, explaining the excellent associated prognosis.
NB has a mixture of neuroblastic and mature ganglion cells and cells intermediate in their differentiation. Various immunohistochemical techniques and electron microscopy are used for diagnosis. Immunohistochemical stains recognize neuron-specific enolase (25), synaptophysin, chromogranin A, and neuronal filaments. Electron microscopy reveals neurofilaments, neurotubules, and neurosecretory granules that contain catecholamines.
The most widely accepted system of histologic classification is now the International Neuroblastoma Pathology Committee Classification (INPC) (47), summarized in Table 6.1. It is based on the highly prognostic system developed by Shimada et al. using patient age, the presence of stroma (“rich” or “poor”) and nodularity, the degree of differentiation, and the mitosis-karyorrhexis index (MKI) (48). Differences between the original Shimada classification (48) and the INPC are (1) to subdivide the “undifferentiated” subtype in the former classification into two subtypes of undifferentiated and poorly differentiated in the latter classification, based on the degree of cellular density and the number of tumor cells and MKI (Fig. 6.4); and (2) to change the name of “stroma-rich, well differentiated” in the former classification to “ganglioneuroma, maturing.” The INPC, based largely on the Shimada system, is now being used in protocols worldwide to facilitate comparisons of patient groups. A retrospective analysis of the Children’s Cancer Group (CCG) validated the prognostic value of this system combined with age (49). An additional modification in 2003 clarified the favorable and unfavorable subsets of ganglioneuroblastoma, nodular (50). Finally, by international consensus, it was shown that even if age were taken out of the INPC, diagnostic category, MKI, and grade of differentiation each had independent prognostic significance (29).
Figure 6.4 A: Fine-needle aspirate of neuroblastoma, showing typical Homer-Wright pseudorosettes with neurofibrillary material in the center. B: Differentiating neuroblastoma, Schwannian stroma-poor. C: Neuroblastoma (Schwannian stroma-poor), poorly differentiated subtype, composed of undifferentiated neuroblastic cells with clearly recognizable neuropil. (From Shimada H, Ambros IM, Dehner LP, et al. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer 1999;86(2): 349-363, with permission.) (See color plate.)
Table 6.1 International Neuroblastoma Pathology Committee System
Prognostic Evaluation of Neuroblastic Tumors According to the International Neuroblastoma Pathology Classification (Shimada System)
a Subtypes of neuroblastoma were described in detail elsewhere.
b Rare subtype, especially diagnosed in this age group. Further investigation and analysis required.
c Prognostic grouping for these tumor categories is not related to patient age.
MKI: mitosis-karyorrhexis index; high is >200 per 5000 cells.
From Shimada H, Ambros IM, Dehner LP, et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999;86(2):364-372, with permission.
CLINICAL PRESENTATION AND EVALUATION
The clinical presentation of NB depends on the site along the sympathetic nervous system chain from which the primary tumor develops (Fig. 6.2) and on the manifestations of metastatic disease. This varies according to patient age. The abdomen is the most common primary tumor location (50-80% of cases). Intra-abdominal primary tumors arise in the adrenals or in a paraspinal location. The paraspinal tumors may have a dumbbell configuration wherein tumor extends through the neural foramina and presents with a mass lesion in the spinal canal; extradural spinal cord compression may occur (51,52). Extra-abdominal locations of the primary tumor include sympathetic ganglia in the neck (often initially thought to be lymphadenitis), posterior mediastinum, and pelvis.
The earliest symptoms or signs of NB may be a palpable abdominal mass (often large, firm, irregular, and crossing the midline), a unilateral neck mass often causing the Horner syndrome, spinal cord compression, respiratory compromise caused by thoracic disease or hepatic metastases placing upward pressure on the diaphragm, or bowel and bladder disturbances caused by compression from a pelvic mass. About 60% of children with NB have metastatic disease (either lymphatic or hematogenous) at the time of clinical presentation. The symptoms in this setting are those of systemic illness: fever, weight loss, weakness, or a general failure to thrive. In a few patients, the presenting symptoms are related to secretory products of the tumor. For example, intractable diarrhea (from vasoactive intestinal polypeptide) may rarely occur, usually in children with ganglioneuroblastoma or ganglioneuroma. In another rare paraneoplastic syndrome, seen in about 3% of patients, a child with NB presents with the opsoclonus (rapid multidirectional eye movements)-myoclonus-truncal ataxia syndrome. This paraneoplastic syndrome is associated with early-stage NB; it is probably caused by development of antineuronal antibodies, and neurologic deficits usually persist despite cure of the tumor (53,54). Bone metastases present as pain, refusal to walk, skull masses, or proptosis with orbital ecchymosis. Skin metastases are common in neonates and generally have a blue tinge (the “blueberry muffin” sign). When a skin metastasis is manually compressed, one may observe blanching of the surrounding skin secondary to liberation of vasoconstrictive catecholamine. NB may infiltrate the marrow and cause pancytopenia with resulting complications (infection, pallor, lethargy, bleeding). Intracranial metastatic disease usually is meningeal, and in infants it may cause cranial suture separation.
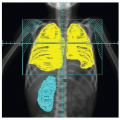
The standard diagnostic evaluation of NB (Table 6.2) includes imaging and laboratory testing with the goals of determining disease extent and prognosis and identifying markers of disease activity. The primary tumor typically is imaged radiographically and with computed tomography (CT) scan or magnetic resonance imaging (MRI). The classic radiographic sign of adrenal NB is calcification in a suprarenal soft tissue mass. In the thorax, a plain radiograph demonstrates a posterior mediastinal mass. In both abdomen and chest, CT scan and MRI are helpful in assessing possible lymph node metastases and intraspinal extension (55).
Table 6.2 Clinical Evaluation of Neuroblastoma (International Neuroblastoma Staging System’s Minimum Recommended Studies to Determine the Extent of Disease)
Tumor Site
Tests
Primary
CT, ultrasound, or MRI with 3D measurements
Metastases
Bilateral posterior iliac bone marrow aspirates and core biopsies (four adequate specimens necessary to exclude tumor)
Bone radiographs and scintigraphy by 99mTc-diphosphonate, with or without 131I-MIBG or 123I-MIBG
Abdominal and liver imaging by CT, ultrasound, or MRI
Chest radiograph (anteroposterior and lateral) and chest CT
Markers
Urinary catecholamine metabolites (vanillylmandelic acid and homovanillic acid)
CT, computed tomography; MIBG, metaiodobenzylguanidine; MRI, magnetic resonance imaging.
From Brodeur GM, Pritchard J, Berthold F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11(8):1466-1477; and Brodeur GM, Seeger RC, Barrett A, et al. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol. 1988;6(12):1874-1881, with permission.
The search for distant, usually bony metastasis must include scanning with 123I-metaiodobenzylguanidine (MIBG) or a technetium (Tc) radionuclide bone scan (for patients whose tumor does not take up MIBG), with conventional radiographs as indicated. Bone metastases are most often periorbital, metaphyseal, and axial in location (Fig. 6.5). Bone scintigraphy is more sensitive than conventional radiographs but is sometimes difficult to interpret in infants younger than 1 year. MIBG is a guanethidine derivative that is similar in structure to norepinephrine and epinephrine. The compound is taken up via the norepinephrine transporter into catecholaminergic cells and stored in the chromaffin granules. It is highly sensitive and specific for skeletal and soft tissue metastases of NB, taken up in more than 90% of primary and metastatic tumors (56). The MIBG is labeled with 123I and scintigraphy is performed for imaging. Because MIBG depends on functional uptake by tumor cells, it is also useful in distinguishing residual active tumor after treatment from masses composed of scar tissue and may complement CT scans (Fig. 6.6) (56). More recently, early response by MIBG scan has been shown to be a useful prognostic marker of response and survival (57). 99mTc-diphosphonate bone scans continue to be used to detect metastatic foci in the skeleton, in patients who are negative by MIBG. Positron emission tomography scans with 18F-fluorodeoxyglucose are a newer modality that may complement MIBG for determination of tumor viability. Several studies have demonstrated that positron emission tomography scanning can visualize NB, even in some cases where MIBG is negative, although in other cases the converse is true (58, 59, 60).
Figure 6.5 Femoral metastases from neuroblastoma.
Liver involvement may be evaluated by CT, ultrasound, or MIBG or PET scan in older children because it is usually focal or nodular. In infants it may be diffuse and not apparent by imaging. For this reason, some authorities recommend liver biopsies for diagnostic workup of infants. Pulmonary parenchymal metastases rarely occur at diagnosis in NB, although at relapse they may be seen in 7% of cases and should be sought with CT scan (61). At diagnosis, lung metastases are associated with MYCN amplification and high lactic dehydrogenase (LDH), and predict a worse outcome (62).
There may be extensive involvement of the bone marrow by tumor without a change in the peripheral blood counts, with some bone marrow tumor present at diagnosis in 80-90% of children with metastatic disease (61). Therefore, bilateral bone marrow aspiration and biopsy are performed routinely, with some centers using 4-10 core biopsies to minimize sampling error. Small amounts of NB cells may be difficult to distinguish from hematopoietic elements. The diagnosis of NB in the bone marrow can be confirmed when only small amounts of tumor are present with immunohistochemical stains on the core biopsy, often using neuron-specific enolase (NSE) as a marker, or with immunostaining on a concentrated cytology specimen from the aspirate. This technology clearly is more sensitive than conventional analyses, detecting one tumor cell per 105 to 106 normal mononuclear marrow cells. The extent of tumor involvement detected by immunocytology at diagnosis in marrow or peripheral blood provides prognostic information (63,64). At present, tumor detected by immunocytology that is not extensive enough to be diagnosed by conventional bone marrow microscopy does not affect staging (55). Reverse transcriptase polymerase chain reaction (RT-PCR) is being used in a number of prospective studies to further quantify the prognostic impact of minimal residual disease (MRD) detected in blood or bone marrow (65,66). Standardized methods for both immunocytology and RT-PCR have been agreed upon by international consensus (67).
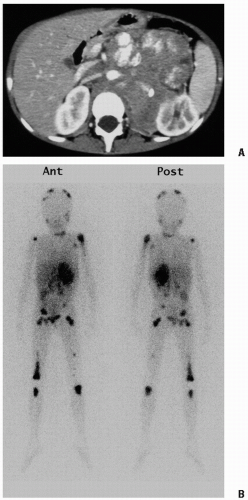
Figure 6.6 A 4-year-old boy presented with fever, abdominal pain, and leg pain. A: The computed tomography scan shows a large retroperitoneal neuroblastoma that encases the abdominal aorta and displaces the inferior vena cava. The left kidney is pushed laterally, and calcifications are evident. B: This 123I-metaiodobenzylguanidine scan shows intense localization in the primary retroperitoneal mass. Widespread bone metastases are evident, with large lesions in the distal right femur and proximal left humerus, but multiple other lesions are seen on the dome of the skull, right humerus, right rib, pelvis, and both femurs and tibias, and in a few retroperitoneal areas, probably representing nodal spread. Physiologic uptake is seen in the salivary glands, liver, and thyroid gland, which was insufficiently blocked with potassium iodide drops.
NB is associated with elevated or abnormal production, secretion, and catabolism of catecholamines (or metabolites) in 90% of cases. Catecholamines and metabolites may be measured in the urine: norepinephrine, VMA, 3-methoxy-4-hydroxyphenylglycol, or HVA. Dopamine may be measured in urine or serum. Urinary catecholamines typically are presented as ratios to urinary creatinine. The symptoms associated with excess catecholamine production include flushing and sweating, pallor, headache, and hypertension.
STAGING AND PROGNOSTIC FACTORS
The criteria for diagnosis of NB recommended by the second International Neuroblastoma Staging System (INSS) Conference are unequivocal pathologic diagnosis from tumor tissue by light microscopy, with or without immunohistology, electron microscopy, elevated urine or serum catecholamines or metabolites (dopamine, HVA, or VMA greater than 3.0 SD above the mean per milligram creatinine for age); or bone marrow aspirate or biopsy containing unequivocal tumor cells (e.g., syncytia or immunocytologically positive clumps of cells) and elevated urine or serum catecholamines or metabolites (55). A diagnosis of NB based only on compatible radiographic findings and elevated urinary catecholamine metabolites is insufficient because of possible confusion with ganglioneuroma or pheochromocytoma or even other solid tumors (e.g., primitive neuroectodermal tumor, rhabdomyosarcoma), which can have false positive urinary findings.
Staging
Four systems, summarized in Evans et al. (68), have been used in previous years for the staging of NB: the international tumor, node, and metastasis system (rarely used in pediatric tumors); the definition by Evans et al. adopted by the CCG; the surgical staging system of St. Jude’s; and the modification adopted by the POG. Subsequent to 1993, most investigators have used the system internationally developed in 1989 and modified in 1993, the INSS (Table 6.3A), in an attempt to improve and unify the previous definitions (55). Most recently, a further modification was introduced by consensus of the International Neuroblastoma Risk Group (INRG) Task Force (69). Understanding the nuances of these systems is important for interpreting and accurately comparing data from various reports and determining therapy.
The INSS was developed in order to have a uniform staging so that studies could be compared, and tried to merge the prior systems of the CCG (Evans) and POG (Table 6.3A). In classifying tumors that cross the midline, infiltration (extending by contiguous invasion to or beyond the opposite side of the vertebral bodies) was chosen to identify tumors presumably less favorable than those that are pedunculated and simply drape over the midline (Table 6.3B). The INSS used Arabic numbers to distinguish it from the letters and Roman numerals of the prior systems. However, some inconsistencies were noted due to the fact that it was a postsurgical staging system with substantial reliance on the assessment of tumor resectability and surgical examination of lymph node involvement. In fact, many patients with stage 3 disease do not actually undergo surgical resection at diagnosis, and many stage 2 patients do not have extensive lymph node sampling, resulting in possibly inaccurate staging. Furthermore, the separation of stage 2A and 2B depending on lymph node involvement is of questionable prognostic value for OS (70). Therefore, four further international conferences were held from 2004 to 2006, and a new international staging system was proposed by the INRG task force in which anatomic staging was performed by radiologic imaging, rather than surgery. In the INRGSS (International Neuroblastoma Staging System), locoregional tumors are staged L1 or L2 based on the absence or presence of 1 or more of 20 image-defined risk factors (IDRFs) (71), respectively. Metastatic tumors are defined as stage M, except for stage MS, in which metastases are confined to the skin, liver, and/or bone marrow in children younger than 18 months of age. Within the 661-patient cohort, IDRFs were present (i.e., stage L2) in 21% of patients with INSS 1, 45% of patients with INSS 2, and 94% of patients with INSS 3 disease. Patients with INRGSS stage L2 disease had significantly lower 5-year event-free survival than those with INRGSS stage L1 disease (78% ± 4% vs. 90% ± 3%; p = 0.0010) (69).
Table 6.3 A: International Neuroblastoma Staging System
Stage
Definition
1
Localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes negative for tumor microscopically
2A
Localized tumor with incomplete gross excision; representative ipsilateral lymph nodes negative for tumor microscopically.
2B
Localized tumor with or without complete gross excision, with ipsilateral lymph nodes positive for tumor; enlarged contralateral lymph nodes must be negative microscopically
3
Unresectable unilateral tumor infiltrating across the midline, with or without regional lymph node involvement; localized unilateral tumor with contralateral regional lymph node involvement; or midline tumor with bilateral extension by infiltration (unresectable) or lymph node involvement
4
Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, and other organs (except as defined for stage 4S)
4S
Localized primary tumor (as defined as stage 1, 2A, or 2B), in patient <1 year, with dissemination limited to skin, liver, or bone marrow (marrow involvement should be minimal with malignant cells <10% of total nucleated cells)
Based on Brodeur GM, Pritchard J, Berthold F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11(8):1466-1477; and Brodeur GM, Seeger RC, Barrett A, et al. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol. 1988;6(12):1874-1881.
Table 6.3 B: International Neuroblastoma Risk Group (INRG) Staging System
Stage
Description
L1
Localized tumor not involving vital structures as defined by the list of image-defined risk factors and confined to one body compartment
L2
Locoregional tumor with presence of one or more image-defined risk factors
M
Distant metastatic disease (except stage MS)
MS
Metastatic disease in children younger than 18 months with metastases confined to skin, liver, and/or bone marrow (< 10%)
From Monclair T, Brodeur GM, Ambros PF, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG task force report. J Clin Oncol. 2009;27(2):298-303, with permission.
International Neuroblastoma Response Criteria (INRC) from the first international conference are still generally used, shown in Table 6.4, which takes into account some of the unique characteristics of this tumor, such as MIBG uptake, catecholamine excretion, and propensity for bone and bone marrow involvement. This is currently the best accepted method for reporting response to treatment, which supplements the National Institutes of Health criteria generally used for evaluating response in phase I and II studies, the Response Evaluation Criteria in Solid Tumors Group (72). As recurrence in NB is most often in metastatic sites in bone and bone marrow, it is critical to be able to evaluate response in these sites in addition to solid masses. To improve the quantization of response in bone sites, a semi-quantitative MIBG score is now used (56).
Table 6.4 International neuroblastoma Response Criteria
Response
Primary Tumor
Metastatic Sites
CR
No tumor.
No tumor; catecholamines normal.
VGPR
Decreased by 90-99%.
No tumor; catecholamines normal; residual 99Tc bone changes allowed.
PR
Decreased by >50%.
All measurable sites decreased by >50%. Bones and bone marrow: number of positive bone sites decreased by >50%, no more than 1 positive bone marrow site allowed.
MR
No new lesions; >50% reduction of any measurable lesion with <50% reduction in any other; <25% increase in any existing lesion.
NR
No new lesions; <50% reduction but <25% increase in any existing lesion.
PD
Any new lesion; increase of any measurable lesion by >25%; previous negative marrow positive for tumor.
CR, complete remission; VGPR, very good partial response; PR, partial response; MR, mixed response; NR, no response; PD, progressive disease. Based on Brodeur GM, Pritchard J, Berthold F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11(8):1466-1477.
Prognostic Factors
Several clinical and biologic characteristics of NB are associated with prognosis. Various cooperative groups and investigators have attempted to select certain variables and group them to identify patients with good, intermediate, or bad prognoses. The differences in various studies probably result from differences such as patient characteristics, treatment, study endpoints. As detailed genomic data are accumulated and interpreted, genetic subsets of NB that reflect clinical behavior are being identified (24,34,73,74). A study of an international database of 8800 NB patients by the INRG showed that patients could be stratified into four different risk groups for 5-year event-free survival (EFS) using age, stage, histologic category, grade of differentiation, MYCN, 11q aberration, and ploidy. The lowest group had an estimated 5-year EFS greater than 85%, while the highest risk group had an EFS below 50% (29). However, for current practical risk stratification on cooperative clinical trials, the most useful and independent variables are stage, age, histology, ploidy, MYCN copy number, and loss of heterozygosity for 1p or 11q. The prognostic values of histology, ploidy, and genetic aberrations have already been discussed above.
Clinical
Stage and age continue to be extremely important determinants of outcome, as originally reported by Breslow and McCann (75). The disease stage strongly influences prognosis (Fig. 6.7). With surgery as primary therapy, more than 90% of children with stage 1 and 2 disease survive and 85% of children with stage 4S survive, whereas with multimodality therapy, the survival is 60-90% for INSS stage 3 disease but only 40% for stage 4 disease (70,76, 77, 78, 79). However, age at diagnosis is another strong determinant of outcome, which must be factored in with stage. Originally 12 months was recognized as the cut-off for a favorable outcome, even in advanced disease, but more recently it has been shown that this can be extended to 18 months as long as the biology of the tumor is favorable. Infants younger than 18 months of age do better than older children with the same disease stage, an effect that is dramatic for patients with advanced stage 3 and 4 disease (37,76,80, 81, 82, 83). Infants and toddlers without MYCN gene amplification and with stage 4 disease have more than 80% long-term survival with modest chemotherapy, whereas children older than 18 months at diagnosis with stage 4 have less than 40% survival with very intensive therapy. This may be related to a higher rate of spontaneous tumor regression or tumor maturation in infants (84). On the other hand, adults and adolescents with NB have a particularly poor long-term survival, even with disease presenting without metastases, although their course may be prolonged (85,86).
Initial response to therapy is another important clinical prognostic factor for patients with advanced disease. Thus, early response by MIBG scan after two to four cycles of induction therapy for stage 4 disease has been shown to correlate with event-free survival (87). In addition, response at the end of induction therapy, prior to myeloablative consolidation is also prognostic of EFS (88, 89, 90).
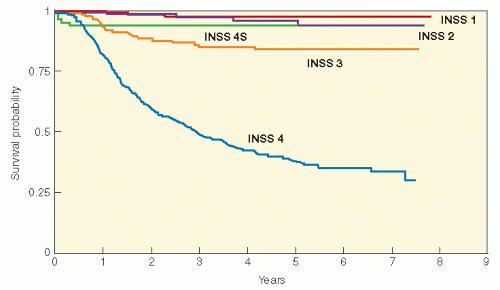
Figure 6.7 Overall survival according to International NB Staging System (INSS) stage of 1034 children with neuroblastoma treated on Children’s Cancer Group protocols from 1991 to 1995. INSS stage 1, n = 195; INSS stage 2, n = 117; INSS stage 3, n = 184; INSS stage 4S, n = 64; INSS stage 4, n = 474. (Courtesy of the Children’s Cancer Group Statistical Office.)
Biologic
Several nonspecific inflammatory serum markers have prognostic value in NB, including ferritin, LDH, and neuron-specific enolase (NSE). All the three are more likely to be elevated in advanced disease and are predictive of a worse outcome in univariate analysis. Ferritin may be produced by NB cells and thus reflects tumor burden or growth, or it may be necessary for NB cell growth. An elevated serum ferritin level (more than 143 ng/mL) is found in up to half of patients with advanced-stage disease but rarely in children with localized disease. PFS is lower for children with elevated levels (91). Neuron-specific enolase is a glycolytic enzyme found in neurons; serum levels greater than 100 ng/mL are associated with lower survival (92). Neuron-specific enolase may be useful as a marker for following the response to treatment. LDH may reflect tumor cell burden or turnover, and serum elevations (750-1500 IU/mL) have been found in some studies to be independently associated with a worse prognosis (93).
Two neural markers found in the serum of NB patients may also be markers of disease activity and prognosis. Ganglioside GD2 is a sialic acid-containing glycosphingolipid found mainly in the cell surface membrane of >95% of NB tumors. Elevated circulating levels may be another marker of disease activity and response to treatment (favorable prognosis level is less than 103 pmol/mL; unfavorable prognosis level is greater than 568 pmol/mL) (94). Chromogranin A is an acidic protein co-stored and co-released with catecholamines from storage vesicles. Its serum concentration is elevated in patients with peptide-producing endocrine neoplasia. The survival rate for patients with lower serum chromogranin A levels (less than 190 ng/mL at the time of diagnosis) was 69%, whereas it was 30% for those with higher chromogranin A levels (95).
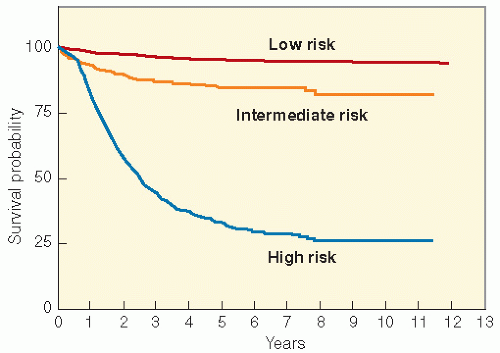
In summary, numerable clinical, biochemical, and genetic risk factors have been shown to have prognostic value in various studies, but the recent multivariate analysis by the INRG has shown that for practical purposes patients can be effectively placed into risk groups for therapeutic assignment using the readily available clinical and genetic factors. Those that have appeared to be consistent over many years and readily available to the clinician have been combined to form a framework for risk stratification in current therapeutic studies. Age, INSS stage, histopathology, tumor cell ploidy, MYCN gene copy number, LOH 1p and 11q are the factors used in risk stratification and therapy assignment for the current COG clinical trials in North America, with very similar guidelines used internationally (Table 6.5). The survival for low-risk patients by this classification is more than 90%, that of intermediate-risk patients is more than 80%, and that of the high-risk group remains at about 30-40% (Fig. 6.8).
Figure 6.8 Survival in 2196 consecutive patients with neuroblastoma treated on Children’s Cancer Group and Pediatric Oncology Group protocols according to clinical risk groups (Table 6.5) assigned according to International Neuroblastoma Staging System stage, age, histopathology, MYCN copy number, and DNA ploidy. Low risk, n = 916; intermediate risk, n = 431; high risk, n = 849. (Courtesy of the Children’s Oncology Group Statistical Office.)
Table 6.5 A: Children’s Oncology Group Neuroblastoma Risk Groups for Treatment
International neuroblastoma staging system
Age (Days)
MYCN
Histology
Ploidy
Risk
1
Any
Any
Any
Any
Low
2A/2B
Any
Nonamplified
Any
Any
Low
Any
Amplified
Any
Any
High
3
<547
Nonamplified
Any
Any
Intermediate
Any
Amplified
Any
Any
High
≥547
Nonamplified
Favorable
—
Intermediate
≥547
Nonamplified
Unfavorable
—
High
≥365 < 547
Nonamplified
Any
Intermediate
4
<547
Nonamplified
Any
Any
Intermediate
Any
Amplified
Any
Any
High
4S
<365
Nonamplified
Favorable
DI > 1
Low
<365
Nonamplified
Favorable
DI = 1
Intermediate
<365
Nonamplified
Unfavorable
Any
Intermediate
<365
Amplified
Any
Any
High
Aberrations of 1p and 11q may be additionally used to adjust therapy intensity, but do not change intermediate-risk patients to high-risk group for outcome.
DI, DNA index.
Table 6.5 B: INRG Risk Classification
INRG Stage
Age (Months)
Histologic Category
Grade of Tumor Differentiation
MYCN
11q Aberration
Ploidy
Pretreatment Risk Group
L1/L2
GN maturing; GNB intermixed
A Very low
L1
Any, except GN maturing or GNB intermixed
NA
B Very low
Amp
K High
L2
<18
Any, except GN maturing or GNB
NA
No
D Low
Yes
G Intermediate
Differentiating
NA
No
E Low
≥18
GNB nodular; Neuroblastoma
Yes
Poorly differentiated or undifferentiated
NA
H Intermediate
Amp
N High
M
<18
NA
Hyper-diploid
F Low
<12
NA
Diploid
I Intermediate
12 to <18
NA
Diploid
J Intermediate
<18
Amp
O High
≥18
P High
MS
NA
No
C Very low
<18
Yes
Q High
Amp
R High
From Cohn Sl, Pearson AD, London WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG task force report. J Clin Oncol. 2009;27(2):289-297, with permission.
SELECTION OF THERAPY
Low-risk tumors are managed with surgery alone unless symptomatic cord compression or respiratory compromise necessitates a short course of chemotherapy. Patients in the low-risk group with stage 1 or 2 disease have an expected 4-year survival of more than 95% with surgery only (70,96), whereas infants with INSS 4S have more than 90% survival with supportive care or a short course of chemotherapy (77). The smaller intermediate-risk group consists of infants and toddlers with stage 4 disease (but no tumor MYCN amplification), favorable biology stage 3, or INSS 4S with unfavorable histology or DNA index. Patients in this group are expected to have an estimated survival of more than 80% with moderate dosages of chemotherapy for 4-8 months and primary tumor resection. Radiotherapy (RT) is rarely used in the low- or intermediate-risk groups because of their favorable prognosis and the fact that many of these patients are younger than 1 year. It is reserved for use in life- or function-threatening situations. It is occasionally needed for cases of unresectable primary disease remaining after chemotherapy or regional recurrences not controlled with chemotherapy.
The high-risk group in NB consists primarily of patients with stage 4 disease who are more than 1 year old at diagnosis but also includes stage 3 with either tumor MYCN amplification or those older than 1 year with unfavorable histopathology, stage 2 above 1 year of age with MYCN amplification, and stage 3, 4, and 4S infants with MYCN gene amplification. Despite the use of increasingly aggressive combined-modality treatments, which have higher remission rates and durations, the long-term survival for INSS stage 4 disease in children who are older than 18 months at diagnosis has remained less than 15% until the early 1990s. The most recent phase III studies indicate that the 3-year EFS of this group has increased in the past decade to 30-40% with the use of myeloablative therapy and treatment for MRD but is still far below the desired outcome (79,90,97). Therapy for high-risk NB is divided into three phases: intensive induction treatment, marrow ablative therapy, and management of MRD. The goal of induction therapy is to achieve maximum reduction of tumor burden, including reduction of bone marrow tumor (in vivo purging), within a timeframe that will minimize the risk of resistant tumor clones and clinical progression. During or at the end of this phase, local control of bulky disease is accomplished with delayed surgery and local RT, which may be given either before or after myeloablative therapy. Subsequently, very high-dose marrow ablative therapy may be used to try to overcome residual and potentially resistant tumor, followed by hematopoietic cell transplant (HCT). The relapse rate of more than 40% even after such treatment (98) has led to the approach of using tumor-targeted therapies after myeloablative treatment to try to eliminate microscopic resistant clones (minimal residual disease).
Chemotherapy
As indicated earlier, chemotherapy is used in low-risk disease only for recurrence or for symptomatic patients with compromised organ function. Intermediate-risk patients can achieve long-term survival with moderate combination chemotherapy for 4-8 months (76,82). The most effective induction regimens for obtaining complete or partial response are combination of platinum-based regimens including other active drugs such as cyclophosphamide, doxorubicin, etoposide, vincristine, and ifosfamide. Induction regimens used in recent large cooperative studies are shown in Table 6.6, with overall response rates generally ranging from about 60% to 80% at the end of 5-6 months of treatment. Most recently, the COG has incorporated two cycles of topotecan with cyclophosphamide into the N7 type induction, with similar response rates and successful peripheral blood stem cell harvests. The European group has tested a dose-intensive schedule in a randomized trial of 262 patients that randomly assigned 132 patients to standard and 130 patients to rapid treatment. The relative dose intensity of the rapid regimen was 1.94 compared with the standard regimen. Myeloablation was given a median of 55 days earlier in patients assigned to the rapid regimen. Although OS did not differ, the 5-year EFS was 18.2% in the standard group and 30.2% in the rapid group, representing a difference of 12.0% (1.8-22.3), p = 0.022 (104). However, the EFS in this study is apparently not different from other recent protocols using less dose intensive regimens. Most of these regimens also include surgery for residual disease, although the overall impact of complete resection on survival in stage 4 disease is still contradictory. Some newer single agents have also been effective in newly diagnosed NB, using the “upfront phase II window” approach. After two courses of single-agent therapy before induction treatment, response rates for effective agents (more than 30% response) were easily detectable, including ifosfamide, carboplatin, iproplatin (99), and topotecan (105). Two agents that were less effective in this setting were epirubicin and paclitaxel. There was no evidence that such a design adversely affected the subsequent outcome of patients when compared with the outcome of patients treated with similar induction without the phase II window.
Myeloablative Therapy with Hematopoietic Stem Cell Support
Myeloablative high-dose chemotherapy with or without total body irradiation (TBI) has been incorporated as consolidation treatment for high-risk NB for the past two decades, beginning with early studies using melphalan ablation. Numerous single-arm studies using varying myeloablative regimens from 1985 to about 1995 showed an apparent improvement in EFS, usually reported from the time of transplantation. The 3-year EFS ranged from 26% to 62%, with an average of about 40% for the large studies (more than 20 patients) shown in Table 6.7 (89,98,106, 107, 108, 109, 111,113,114). This approach introduced some bias into the interpretation of results because only patients who survived for 5-6 months and responded to induction chemotherapy were able to receive the high-dose myeloablative treatment with autologous or allogeneic transplantation.
Only gold members can continue reading. Log In or Register to continue