2.8: Nonspecific inflammatory disorders
Sinduja Sivaramalingam, Alok Kale, Pradhap Lenin, N. Chidambaranathan
Some of the uncommon disorders with multisystem manifestations do not have a singular cause or well-proven pathogenesis and do not fit into one specific group. Variable mutations, immune dysregulation, inflammatory cascade and other unknown causes, in isolation or in combination, may be the contributory factors for these disorders. For the sake of convenience, such pathologies are dealt, in this chapter.
Histiocytic disorders have predominant proliferative characteristics with multifocal involvement. Other proliferative disorders such as Castleman’s disease, Kikuchi and Kimura’s disease involve lymph nodes. Cystic fibrosis is a channelopathy with significant inflammatory component.
These diseases have variable clinical course based on the number of sites or system of involvement. While unifocal involvement involving noncritical areas has good outcome, multisystem and disseminated course of the diseases have poor prognosis.
Role of imaging in nonspecific disorders
Most of the aforementioned proliferative disorders have nonspecific imaging features. Hence, histopathological examination with immunotyping remains the gold standard. Nevertheless, typical lesion in typical location with supportive clinical information can point to the diagnosis or at least raise a concern to consider or add it in the differentials.
PET-CT and scintigraphic studies can be immensely helpful from the screening point of view, to look for other sites of involvement and also in follow-up. Follow-up is recommended for evaluation of treatment response and for surveillance of metachronous lesions.
For characterization of individual lesions or assessment of a particular high-risk region, modalities with superior resolution such as CT or MRI with contrast are recommended.
In-depth working knowledge of a radiologist can prove to be greatly beneficial in the evaluation of these pathologies. Also, it is desirable to set a well-tailored imaging protocol, based on the patient configuration and the institutional constraints in every tertiary care setting, so as to guide in the management of these disorders.
HISTIOCYTIC DISORDERS
Sinduja Sivaramalingam, N. Chidambaranathan
Introduction to histiocytosis
Histiocytosis is an “umbrella” term encompassing a rare group of disorders characterized by proliferation and accumulation of various types of histiocytes (of macrophages, dendritic cells, monocyte lineage) in various tissues and organs of children and adults.
Some of the histiocytic disorders like the sarcomas and leukaemias are clearly malignant. The pathogenesis behind other histiocytic disorders has long remained enigmatic with two contrasting theories. The advent of specific somatic oncogenic mutations, the associated monoclonal or oligoclonal cellular proliferation and favourable response to chemotherapeutic drugs have tilted the balance towards the neoplastic origin theory in most of these disorders. The polyclonal proliferation of cells in specific conditions like adult onset pulmonary LCH which show good response to withdrawal of the inciting insult, namely cigarette smoke, points towards the reactive or inflammatory process theory.
The classifications for histiocytic disorders are pathology-based, exhaustive and complex (Tables 2.8.1.1, 2.8.1.2). From the clinical and radiological standpoint, the proliferative histiocytic disorders can be further divided into langerhans cell histiocytosis (LCH) and non-langerhans cell histiocytosis. Some of the non-LCH disorders to be described in this chapter are Erdheim–Chester disease (ECD), Rosai–Dorfman disease (RDD), Juvenile XanthoGranuloma (JXG) and Haemophagocytic LymphoHistiocytosis (HLH).
Reproduced from: J.F. Emile, O. Abla, S. Fraitag, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 127 (22) (2016) 2672–2681. doi:10.1182/blood-2016-01-690636.
Reproduced from: B.E. Favara, A.C. Feller, M. Pauli, E.S. Jaffe, L.M. Weiss, M. Arico, et al. Contemporary classification of histiocytic disorders. Med. Pediatr. Oncol. 29 (3) (1997) 157–166.
General features of histiocytic disorders
- • Isolated or multifocal, multisystem course: Proliferative histiocytic disorders may show either isolated single-system involvement or multifocal, multi-systemic disease. Unifocal lesions are sometimes self-regressing and have a good prognosis. Multifocal and multisystemic disseminated disease patterns have guarded prognosis.
- • Overlapping and non-specific imaging features: Can be seen between the different types of histiocytic disorders.
- • Importance of associated factors: In view of non-specific imaging features, due importance should be given to the nature of other concomitant lesions, age at presentation, histological features, clinical presentation, response to therapy. These points play a critical role in excluding each disease from the exhaustive list of differentials.
- • The gold standard: Histopathological examination coupled with immunohistocytology is the gold standard for diagnosis. The tissue sample if possible is obtained from the most accessible sites like skin and subcutaneous soft tissue. Bone and visceral biopsies are done as the next resort.
- • Multimodality imaging plays a principal role in the evaluation of these histiocytic disorders. Modalities like Radiography, Tc-99 scintigraphy and PET-CT are meant for whole body survey for multisystem lesions, follow up and for assessing response to therapy; whereas CT and MRI focusses on intricate characterization of the individual lesions or a particular system.
- • Need for multisystem imaging: Some of the proliferative lesions in critical areas or organs maybe asymptomatic at presentation. Without multisystem screening, these lesions may be left undetected, may progress to present later to cause significant mortality and morbidity in the advanced phase of the disease. It is advisable that each department follow an optimal imaging protocol for multisystem screening, based on the available resources, age and clinical presentation.
- • Role of follow-up: Lesions in the histiocytic disorders can be synchronous or metachronous. Hence follow-up imaging plays cardinal role not only assessing the treatment response but also in surveillance of the other system to check for new lesions.
Langerhans cell histiocytosis
Introduction
Langerhans cell histiocytosis (LCH) or histiocytosis X is a rare disorder characterized by clonal proliferation of a special type of immature dendritic cell (DC) called the Langerhans type of histiocytes, infiltrating different organs.
Epidemiology
Most often recognized in childhood, between 1 and 15 years old, it has a peak incidence between 5 and 10 years of age. Slight male preponderance is documented.
Histopathological features in LCH
Epidermal Langerhans cell, first described by Paul Langerhans in 1868, is a tissue resident dendritic cell, present in the skin (especially stratum spinosum), bronchi, mucosa, etc. They play a key role in antigen presentation and have intracytoplasmic organelles known as Birbeck granules. Birbeck granules have a pentalaminar, rod like, tubular appearance with tennis-racket appearance.
Due to the structural similarity between the pathological cells described in LCH and the epidermal langerhans cells, they were termed as Langerhans cells histiocytosis. Studies later showed that these LCH cells are basically different, with direct myeloid origin (contrary to the belief that they are products of abnormal differentiation of the epidermal langerhans cells). They play no role in antigen presentation, have different surface antigens and are capable of infiltrating various organs that do not normally contain the epidermal Langerhans cells. LCH lesions are essentially granulomatous, containing cellular infiltrates of langerhans cell histiocytes with a variable background of inflammatory cells and multinucleated Giant cells. Immunologically LCH cells express HLA-DR, S-100, langerin and CD1a.
The term histiocytosis X was termed by Lichtenstein in 1953, after observing the common histologic appearance of lesions in varied syndromes (like Hans–Christian Schuller, Letterer Siwe disease, Eosinophilic granuloma), the pathology of which were till then unknown.
Etio-pathogenesis and associated mutations
Somatic mutations in the BRAF V600E oncogene are identified in almost 50% of LCH patients. Because of the aberrant protein, there is dysregulation of the RAS/RAF/MEK/ERK signalling pathway, which in turn contributes to the uncontrolled proliferation of the Langerhans cells. BRAF V600E mutation in LCH is associated with multisystem involvement with severe clinical course, resistance to chemotherapy and an increased risk of relapse.
The next common mutation involves the MAP2 K1 or MEK1 and is identified in 25% of the cases.
With the evolving understanding that somatic mutations leads to pathological dys-differentiation of Monocyte–Phagocyte system, LCH is now considered to be an “inflammatory myeloid neoplasia”.
Other factors like viral infections and environmental toxins are also believed to contribute to the development of this complex disorder.
LCH is generally a sporadic, non-hereditary condition. However, rare familial occurrence has been reported, particularly in identical twins.
Studies show a recognized association between LCH, haematological and solid malignancies.
Common genetic etiology has been identified for Langerhans cell histiocytosis (LCH), acute myeloid leukaemia (AML) and primary idiopathic myelofibrosis (MF).
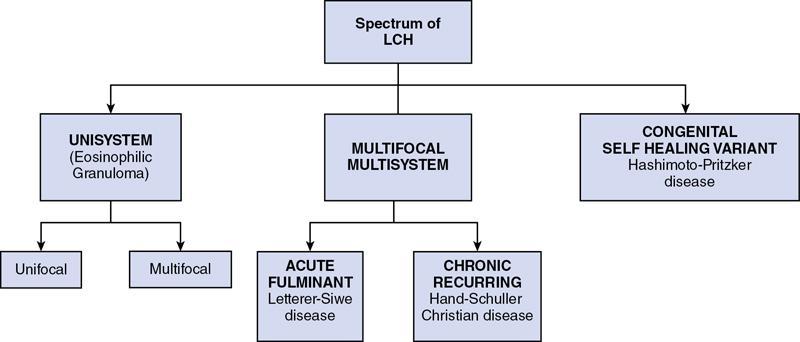
Spectrum of LCH
LCH is known to virtually affect any organ of the body, frequently affected organs are bones (80%), skin (33%) pituitary gland (25%), liver, spleen, haematopoietic system or lungs (15% each), lymph nodes (5%–10%) or the central nervous system (CNS) (2%–4% excluding the pituitary).
The clinical manifestation of LCH depends on its severity and the number of organs involved and ranges from single-site, self-limited lesions to fatal disseminated disease.
Unisystem – unifocal and multifocal langerhans cell histiocytosis
Eosinophilic granuloma is characterized by Langerhans cells proliferation involving a single system – most commonly the bones, rarely involving the skin or lungs.
(A) Unifocal unisystem lesions or the localized form of LCH is the most common (∼70%) pattern usually affecting the skeletal system in older children or adults between 5 and 15 years of age. The lesions are limited to a single bone or a few bones, and may involve the lung.
They can be asymptomatic, or present with pain, tenderness or pathologic fractures. As an indolent disorder, it can heal spontaneously or be cured by local excision or irradiation.
(B) Multifocal unisystem disease or the chronic recurring form of LCH (comprising ∼20%) usually affects young children, typically between 1 and 5 years of age (earlier than those with unifocal disease). This pattern presents with multiple bone lesions and involvement of the reticuloendothelial system (i.e. the liver, spleen, lymph nodes and skin). When the pituitary gland is involved, it leads to diabetes insipidus in half the patients.
Multifocal multisystem langerhans cell histiocytosis
(A) Letterer–Siwe disease, the acute and most fulminant form, constituting about 10% of the LCH cases, frequently affects children below 2 years of age.
This usually starts as cutaneous lesions similar to seborrheic eruptions in the front, back of the trunk and on the scalp. In addition, hepatosplenomegaly, lymphadenopathy, pulmonary lesions and multiple destructive osseous lesions also occur as a part of the systemic process. Extensive bone marrow infiltration causes anaemia, thrombocytopenia and in turn predisposes to recurrent infections such as otitis media and mastoiditis. It runs a fulminant course and is notoriously fatal within 1–2 years. Even with intensive chemotherapy, the prognosis is guarded, with only 50% of patients surviving over 5 years.
(B) Hand–Schuller–Christian disease is a chronic recurring variant of LCH affecting children of 1–5 years age group. It consists of multiple osseous lesions with extraskeletal multisystem involvement. The combination of calvarial bone defects, diabetes insipidus, and exophthalmos is referred to as the Hand–Schuller–Christian triad. Many patients experience spontaneous regression; others can be treated successfully with chemotherapy.
Diagnosis of LCH and multisystem screening
Diagnosis of LCH is made by histopathological examination, with tissue obtained from easily accessible sites like skin, soft tissue (Table 2.8.1.3). Rarely bone biopsy is performed. Assessment of endocrine functions is also done when Hypothalamo–Pituitary axis is involved.
| Histopathological examination specimen | Tissue biopsy, bone biopsy |
| Haematoxylin-eosin stain | Features of Langerhans cells – like abundant, vacuolated eosinophilic cytoplasm, distinct cell margins |
| Electron microscopy | Presence of Birbeck granules |
| Immuno-histo-chemistry (IHC) | CD1a, langerin, S-100 positive |
When an LCH lesion is diagnosed in a child, a radiographic skeletal survey is recommended to look for other lesions. Though radiography is considered the modality of choice for skeletal survey in view of availability and easiness, its sensitivity is questionable. An osteolytic lesion in a radiograph becomes evident only when 30%–50% of the bone destruction has already occurred. Moreover lesions in scapula, skull base and other regions overlapped by other bones or bowel shadow can be missed if we depend on radiograph alone.
Whole body MRI is considered to be advantageous for assessing the soft tissue component of the disease, precise marrow involvement and especially extra-osseous lesions. However, the cumbersome protocol, duration and the need for patient co-operation, especially in paediatric patients, question its validity as a screening modality, in day-today practise.
Bone scintigraphy is another important tool in lesion screening; however, the sensitivity and specificity of the modality is relatively low.
FDG PET-CT is another newer modality with the capability to provide excellent morphological and functional data. It has established a solid role in the follow up of multisystem diseases. But the amount of radiation exposure, especially for the paediatric patients questions its frequent use.
Clinical presentation, radiological features, differentials and management
LCH frequently involves the skeletal system in 80% of the cases. Other sites of involvement are skin (33%), pituitary (25%), haematopoietic system, liver, spleen and lungs (15%), lymph nodes (5%–10%), central nervous system excluding pituitary (2%–4%) and thymus. Other rare sites like breast, thyroid, etc., have also been described in literature.
Skeletal lesions
Skeletal system remains the most commonly affected system (approximately in 80% cases). LCH has a predilection for the flat bones, especially the skull, followed by the mandible, ribs, pelvis and spine.
Unifocal osseous lesions are usually confused with osteomyelitis, because of associated low grade fever, elevated ESR, mild leukocytosis and normochromic anaemia which is present in both these conditions. About 10% of those with localized LCH, proceed to develop multifocal and extraskeletal lesions (Fig. 2.8.1.1).
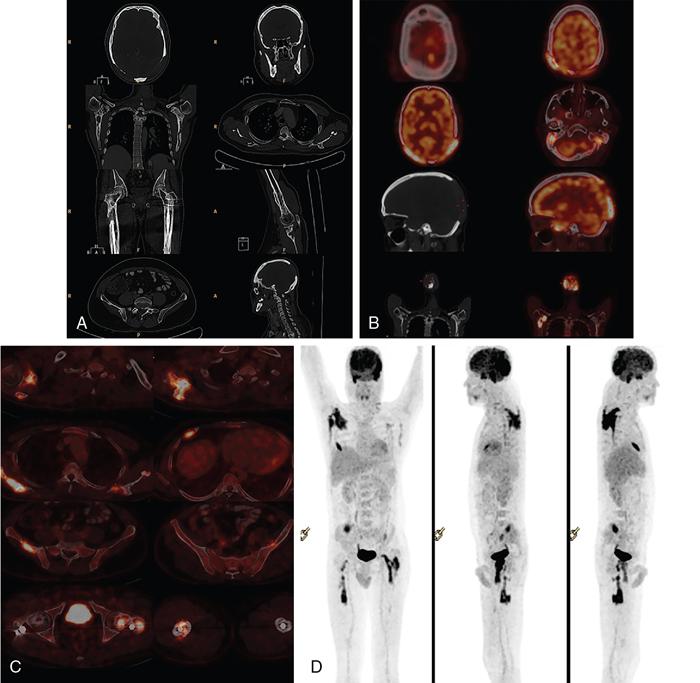
Typically, the soft tissue component of the lesions appears isoechoic at ultrasonography (US).
The signal intensity of the soft tissue component in the MRI depends on the stage of the disease. Active lesions with high cellularity show high signal on T2 weighted images with contrast enhancement, whereas chronic fibrotic lesions tend to show low signal on both T1- and T2-weighted images. Active lesions also show significant associated marrow, periosteal and soft tissue oedema. An endosteal rim of hypointensity can be considered as an early sign of healing (Table 2.8.1.4).
| Active Cellular Lesions | Chronic Fibrotic Lesions | |
|---|---|---|
| T1-weighted images | Hypo to iso-intense | Hypointense |
| T2-weighted images | Hyperintense | Hypointense |
| STIR | Hyperintense | Hypointense |
| T1W + contrast | Avid contrast enhancement | Minimal or no contrast enhancement |
Bone scintigraphy: Active and aggressive lesions show avid radiotracer uptake. Some active lesions may also show peripheral rim of increased radiotracer uptake with central photopenia. Older and quiescent lesions may show no significant uptake.
Management of skeletal lesions
Treatment options depend on the site and extent of the lesions, whether the lesions are single or multiple. Single skull and long bone lesions are treated with curettage with or without intralesional steroid injection or radiation therapy.
Chemotherapy and oral systemic steroids may be used to treat multiple lesions and to prevent CNS extension, when mastoid, orbit are involved.
Calvarium and skull base.
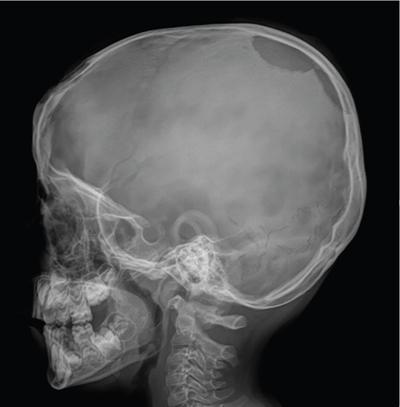
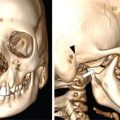
The most common site of bone involvement in LCH is the skull, calvarium in particular. Parietal bone is the most common bone involved in the calvarium. Patients may be asymptomatic, or present with painless swelling or focal scalp pain.
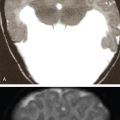
Typically the skull lesions are well-defined, lytic and “punched-out” with asymmetric destruction of the inner and outer cortices giving a “characteristic bevelled edge” (Fig 2.8.1.2). Healing lesions are known to have sclerotic margins (Fig 2.8.1.3). Multiple, lytic skull lesions may coalesce, creating a map-like appearance referred to as a “geographic skull”.
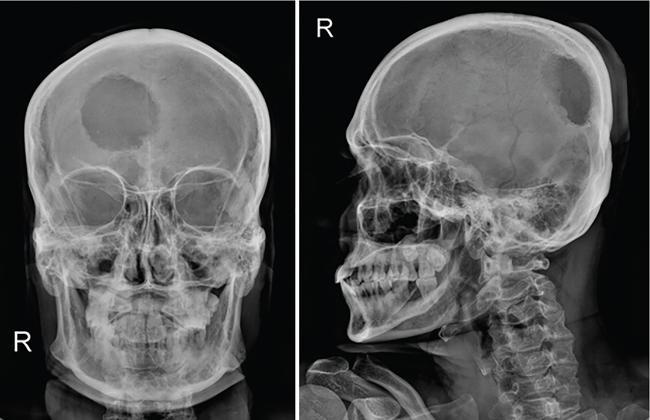
Calvarial lesions lack periosteal reaction unlike other locations.
Another described finding is the “classic button sequestrum or bull’s eye pattern”, with central sclerosis and surrounding radiolucency.
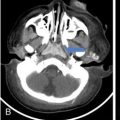
Infrequently the lesions may also cross suture lines. The perforating lytic lesions may show epicranial, transdural involvement with dural tail (Fig 2.8.1.4). Intraxial infiltration of the brain parenchyma with associated oedema is another rare finding.
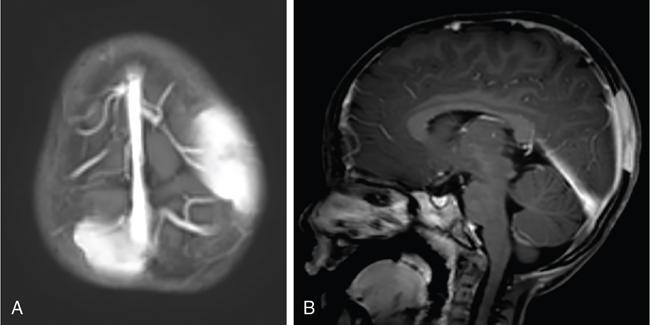
The skull base is frequently involved next to the calvarium, mastoid bone in specific. Mastoid involvement may be unilateral or bilateral and typically presents with swelling, dizziness, vertigo and otorrhea, clinically mimicking mastoiditis.
Squamous portion of the temporal bone and middle ear are less frequently involved. Involvement of clivus, jugular foramen and cranial nerve palsies has been uncommonly reported.
Differential diagnosis for active calvarial lesions includes metastasis, leptomeningeal cysts. Chronic lesions with sclerotic margins may mimic epidermoid and dermoid cysts, meningocele, chronic osteomyelitis, etc. Differentials for multiple lesions include lymphoma, leukaemia, multiple myeloma and metastases.
Other flat bones.
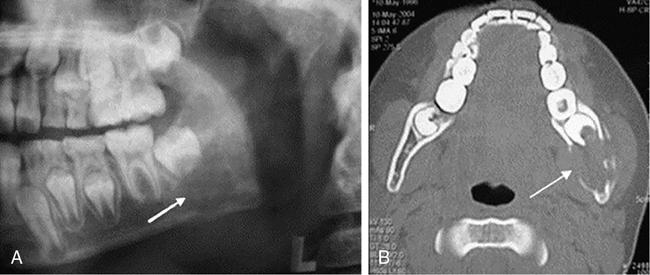
Isolated flat bone involvement is more frequent in adults and typically presents as rib or mandibular lesions. Gnathic or mandibular lesions may be alveolar or intra-osseous in location.
The alveolar LCH mimics periodontitis and presents with pain and gingival bleeding. They give the radiographic appearance of “floating teeth” due to the scooped-out alveolar destruction mostly involving the middle of roots (Fig 2.8.1.5).
I ˙  . Haberal Can, A. Kurt, E. Özer, N. Sarı, E. Samim. Mandibular manifestation of Langerhans cell histiocytosis in children. Oral Oncology Extra 41 (8) (2005), 174–177).
. Haberal Can, A. Kurt, E. Özer, N. Sarı, E. Samim. Mandibular manifestation of Langerhans cell histiocytosis in children. Oral Oncology Extra 41 (8) (2005), 174–177). Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


