potential radiation risks are lower than in the axial skeleton, which is located near more radiosensitive mediastinal, abdominal, and pelvic organs.12 More recently, selective use of fetal CT with 3D reformations has been described in cases in which US and genetic data are inconclusive to either diagnose or exclude a suspected skeletal dysplasia with impact on accurate counseling of families.5,12,13,14,15
 FIGURE 21.1 Normal bone marrow conversion in long bones. Sequential age-related changes in hematopoietic marrow to fatty marrow distribution are illustrated in this figure representing a femur. |
development of a secondary ossification center and progressive conversion into bone.
of long bones, also guides the reparative response to stimuli (e.g., trauma, infection, neoplasm, metabolic disorder, and nutritional status). In children, the periosteum is physiologically more active and less adherent to the cortex than in adults, allowing for earlier and more aggressive appearance of the periosteal reaction.26 The differential diagnosis for benign periosteal reaction in the pediatric population is summarized in Table 21.1.
 FIGURE 21.2 Physiologic genu varum and metaphyseal beaking in a 19-month-old girl. Frontal radiograph of bilateral lower extremities shows symmetric genu varum with medial metaphyseal beaking in both distal femurs (arrows) and proximal tibias (interrupted arrows). Patient was asymptomatic and genu varum resolved 1 year after radiographic evaluation. |
 |
 FIGURE 21.3 Physiologic periosteal reaction in a 2-month-old boy. A: Frontal radiograph of the right tibia. B: Frontal radiograph of the left tibia. Both images show thin, smooth, continuous, symmetric periosteal reaction (arrows) along the medial diaphyseal cortex of both tibias. Included portions of the distal femurs also show physiologic periosteal reaction (interrupted arrows). |
TABLE 21.1 Differential Diagnosis of Physiologic Periosteal Reaction (1-6 Months of Age) | |||||
|---|---|---|---|---|---|
|
 FIGURE 21.4 Cortical desmoid in a 4-year-old boy. A: Frontal radiograph of the right knee shows a lucent lesion (arrow) with well-marginated sclerotic borders in the medial femoral metadiaphysis. B: Lateral radiograph of the right knee shows the typical posterior location with sclerosis and mild irregularity (arrow). C: Axial fat-saturated T2-weighted MR image shows subperiosteal location of the lesion (arrow) with increased signal intensity and sclerotic borders. D: Sagittal fat-saturated intermediate gradient-echo sequence MR image shows homogeneous hyperintense signal intensity (arrow) consistent with fibrous tissue and anatomic relationship to the insertion of the medial head of the gastrocnemius (arrowhead). |
bony abnormalities in the axial and appendicular skeleton of affected pediatric patients. This information should be integrated with the rest of the information provided by a multidisciplinary team approach in order to reach a correct diagnosis. A comprehensive analysis of imaging findings in skeletal dysplasias is beyond the scope of this chapter. However, the most common skeletal dysplasias identifiable through imaging at birth or later in life are summarized in this chapter. In addition, the most common disorders of limb reduction, congenital bowing of the legs, and congenital foot deformities are reviewed. Finally, syndromic skeletal abnormalities, developmental hip dysplasia, and skeletal abnormalities associated with neuromuscular disorders are discussed (Table 21.2).
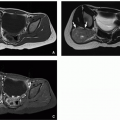
 FIGURE 21.5 Discoid lateral meniscus in a 7-year-old girl with knee pain and locking. Coronal fat-saturated intermediate sequence MR image demonstrates diffuse enlargement of the lateral meniscus (arrow) completely occupying the central portion of the lateral compartment. Linear increased intrasubstance signal intensity is noted along the width of the discoid lateral meniscus (arrowheads); however, there is no meniscal tear extending along the articular surface. Normal size medial meniscus is noted (interrupted arrow). |
TABLE 21.2 Selected Spectrum of Skeletal Dysplasias with Identifiable Imaging Findings at Birth and Later in Life | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
 FIGURE 21.6 Thanatophoric dysplasia in a newborn girl. A: Frontal radiograph of the pelvis demonstrates bilateral small iliac bones with “trident acetabulum” (arrow), shortening and bowing of the femurs “French telephone receiver” (interrupted arrow), and diffuse narrowing of the interpedicular spaces (arrowheads). B: Lateral radiograph of the thoracolumbar spine demonstrates severe platyspondyly (arrows) and short ribs with anterior cupping (interrupted arrows). Short upper and lower limbs are also noted. |
 FIGURE 21.7 Chondrodysplasia punctata in a 5-day-old girl. A: Frontal radiograph of the abdomen demonstrates stippled calcifications in both proximal femoral epiphyses (arrows). B: Lateral radiograph of the right humerus demonstrates similar stippled calcifications in the proximal humeral epiphysis (arrow). |
posterior vertebral scalloping, narrow chest with short ribs, rhizomelia with short thick tubular bones and metaphyseal flaring, notched physis (V shaped), short metacarpals and phalanges in “trident” configuration (separation between third and fourth fingers), horizontal acetabular roof, and squared iliac wings (“tombstone configuration”) (Fig. 21.8). Prenatal US demonstrates short long bones, particularly femurs and humeri, flat vertebral bodies, and large skull. In addition, small chest, short fingers, and polyhydramnios have been described.46
 FIGURE 21.8 Achondroplasia in a 1-year-old girl. A: Frontal radiograph of the hand demonstrates short, broad, long bones of the forearm and hand. There is V-shape configuration of the distal ulnar physis (arrow) and “trident” configuration of the hand. B: Frontal radiograph of the pelvis and lower extremities demonstrates squared iliac bones or “tombstone configuration” (arrow), horizontal acetabular roofs (interrupted arrow), decreased interpedicular space in the lumbar spine (interrupted lines), and short tubular long bones with metaphyseal flaring in bilateral lower extremities. |
the onset of symptoms ranges from infancy to early childhood rather than at birth. The most common types of metaphyseal chondrodysplasia are Schmid, McKusick, and Jansen variants.
 FIGURE 21.9 Metaphyseal chondrodysplasia in a 6-year-old girl. A: Frontal radiograph of the pelvis demonstrates bilateral coxa vara (arrows), enlarged capital femoral epiphyses (interrupted arrow), and physeal widening (arrowhead). B: Frontal radiograph of the right tibia demonstrates physeal widening (arrows) and metaphyseal flaring (interrupted arrows). |
with apex extending away from the joint (Fig. 21.10). The osteochondromas can also be seen in flat bones, hands, ribs, and spine. There is the potential risk for malignant transformation in ˜5% of affected patients with multiple cartilaginous exostoses, most commonly chondrosarcoma.62
 FIGURE 21.10 Multiple cartilaginous exostoses in a 14-year-old boy. A: Frontal radiograph of both knees as part of scanogram examination for leg length discrepancy demonstrates numerous bilateral sessile (arrow) and exophytic osteochondromas (interrupted arrow). Note predominant metadiaphyseal distribution with growth directing away from the joint. B: Frontal radiograph of the left forearm with multiple osteochondromas of the proximal and distal radial and ulnar metadiaphysis. There is characteristic shortening and bowing of the distal ulna (arrow) with medial angulation of the distal radial epiphysis (interrupted arrow). |
 FIGURE 21.11 Maffucci syndrome. This 15-year-old boy had a history of multiple enchondromas, all showing benign lobulated cartilage (left). Biopsy of a vascular lesion with cavernous spaces resembling venous malformation as well as solid areas resembling spindle cell hemangioma (right) confirmed the diagnosis of Maffucci syndrome. (Both hematoxylin and eosin, original magnification, 200×.) |
of an extremity with limping, and osseous deformities with possible pathologic fractures.64
 FIGURE 21.12 Enchondromatosis in an 8-year-old boy with leg length discrepancy. Frontal view of both knees as part of scanogram examination demonstrates a radiolucent lesion with calcific matrix (“ring-and-arc” pattern) in the left distal femoral metadiaphysis (arrow) abutting the physis consistent with a large enchondroma. A subtle linear enchondroma is seen in the left proximal tibial metadiaphysis (interrupted arrow) with tiny regions of chondroid matrix (arrowheads). Shortening of ˜4 cm in the left lower extremity relative to the right secondary to enchondromatosis is also noted (not shown). |
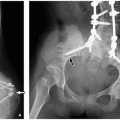
 FIGURE 21.13 Polyostotic fibrous dysplasia in a 9-year-old boy with unilateral distribution in the appendicular skeleton and facial involvement. A: Lateral skull radiograph demonstrates facial deformity secondary to fibrous dysplasia with characteristic ground-glass expansile lesions of the base of the skull and frontal bone, maxilla, and the anterior mandible (arrows). B: Frontal radiograph of the pelvis demonstrates coxa vara deformity with expansile mixed-sclerotic fibrous dysplasia lesion in the right femoral neck (arrow). A similar smaller fibrous dysplasia lesion is noted in the medial aspect of the left proximal femoral diaphysis (interrupted arrow). C: Frontal view of the right humerus demonstrates an expansile ground-glass lesion of the mid humeral diaphysis with subtle endosteal scalloping and cortical thinning (arrow). |
and malignant). Musculoskeletal manifestations of NF1 include skull abnormalities (sphenoid wing dysplasia, erosions, and enlargement of foramina), scoliosis and kyphosis, pseudarthrosis of long bones (tibia most common), osteoporosis, and short stature (Fig. 21.15).72
 FIGURE 21.14 Neurofibromatosis type 1. Plexiform neurofibroma, diffusely expanding a peripheral nerve and its branches (upper panel), is seen. The cut surface (lower left) is tan white and firm, resembling fibrous tissue. In this example, the majority of the mass has the histologic appearance of neurofibroma (lower center), whereas areas of increased cell density and round “epithelioid” cells characterize a component of malignant peripheral nerve sheath tumor (lower right). (Hematoxylin and eosin, original magnification, 400×.)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|