, Ahmad Elkhodary2 and Abdelhamid H. Elgazzar3
(1)
Oncology Center of Excellence, International Medical Center, 2172, Jeddah, 21451, Saudi Arabia
(2)
Department of Medical Oncology, National Cancer Institute, Cairo University, Cairo, Egypt
(3)
Department of Nuclear Medicine, Faculty of Medicine, Kuwait University, Kuwait City, Kuwait
11.1 Tumor Pathology
11.1.1 Biological Behavior
11.1.2 Grading
11.1.3 Tumor Staging
11.1.4 Rate of Growth
11.2 Tumor Biology
11.2.1 Cell Growth and Cell Cycle
11.2.4 Invasion and Metastasis
11.2.5 Carcinogenesis
11.2.6 Apoptosis
11.2.7 Senescence
11.2.8 Hereditary Cancer
11.3 Summary
During the past century, there has been a gigantic evolution in our understanding of the perplexing concepts of the biology of human tumors. This chapter is intended to address only the knowledge that has had the most significant impact in this field. Owing to the constraints of space, this chapter is not to be considered all inclusive. We will deal with the pathological basis of tumorigenesis and the basic principles of pathological classification of tumors.
11.1 Tumor Pathology
The classification and typing of tumors depends mainly on the histopathological diagnosis, which is made on the basis of gross and microscopic examination of tissues. The surgical pathologist’s task is to provide a specific and comprehensive diagnosis to enable the clinician to develop an optimal plan of treatment and help him or her to predict prognosis.
Tumor classification is based on histogenesis, degree of cellular differentiation (i.e., well or poorly differentiated), and biological behavior (benign versus malignant). All tumors, whether benign or malignant, have two components: (1) proliferating neoplastic cells and (2) supportive stroma, which is host derived and made up of connective tissue and blood vessels. While the neoplastic cells determine the nature of the tumors, tumor growth and evolution depend on the stroma [1].
In the past, the general concept was that neoplasms of certain phenotypes arise from their normal cell counterpart. However, evidence that accumulated over the years has proven the inaccuracy of this histogenetic assumption. It is now believed that most tumors arise from immature cells that can transform and acquire phenotypic features similar to those of one or more normal cell types. For example, rhabdomyosarcomas are tumors that show rhabdomyoblastic differentiation rather than tumors that arise from striated muscle cells [2]. In some instances, the immature cells can undergo divergent differentiation into two cell types, as in the case of mixed tumors of the salivary gland (Fig. 11.1), or they have the capacity to differentiate into any adult cell type, as in teratoma.
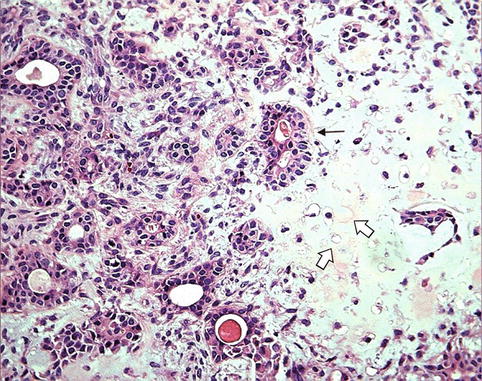
Fig. 11.1
Benign mixed tumor of the salivary gland (pleomorphic adenoma). The tumor consists of an epithelial component “glands” (arrow) and a mesenchymal component “cartilage” (open arrows)
11.1.1 Biological Behavior
The categorization of tumors into benign and malignant is an oversimplification of the wide behavioral range of neoplasms. There are tumors that exhibit intermediate behavior. This has led to the introduction of a third category designated as “borderline or undetermined,” which represents low-grade malignant tumors that can mostly be managed by conservative therapeutic approach. The best examples are borderline tumors of the ovary and uterine smooth muscle of low malignant potential [3–5]. Currently, the malignant category is restricted to tumors that have metastatic properties.
11.1.1.1 Benign Tumors
In general, the addition of the suffix -oma to the cell of origin describes benign tumors; for example, adenoma indicates a benign tumor of epithelial cell origin. Tumors that arise from mesenchymal tissues are designated according to their putative cell of origin (e.g., fibroma, chondroma, lipoma, and leiomyoma). Benign tumors can also be classified on the basis of their macroscopic pattern; for example, papillomas are benign epithelial tumors with certain growth characteristics, such as exophytic or finger-like projections. In general, a benign tumor is composed of well-differentiated cells that resemble their normal counterpart. A tumor is considered benign when its gross and microscopic characteristics are relatively innocent, implying that it will remain localized and cannot spread to other sites. However, it should be noted that benign tumors can produce more than a localized mass and sometimes cause serious disease.
11.1.1.2 Malignant Tumors
The classification of malignant tumors essentially follows that of benign tumors, with some exceptions. Malignant neoplasms arising from epithelial cells are termed carcinomas. Carcinomas are further classified on the basis of the type of epithelium, for example, squamous as squamous carcinoma (Fig. 11.2), glandular as adenocarcinoma (Fig. 11.3), and transitional as transitional cell carcinoma. Malignant epithelial tumors that have not extended through the underlying basement membrane are described as in situ carcinoma. Malignant tumors arising from mesenchymal tissue are broadly designated as sarcomas. These are further subclassified on a histogenetic basis according to the normal tissue they resemble or its embryonal counterparts, for example, fibrosarcoma, chondrosarcoma, leiomyosarcoma, and rhabdomyosarcoma. Lymphomas are tumors of lymphatic system cells, while myelomas are those of plasma cells.
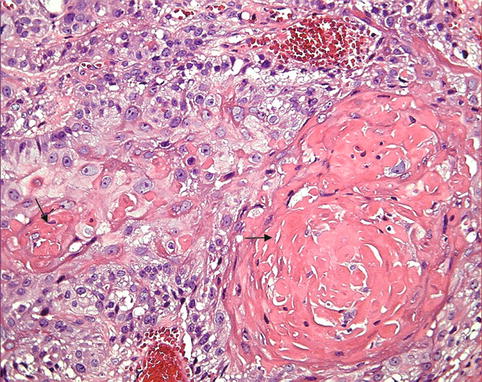
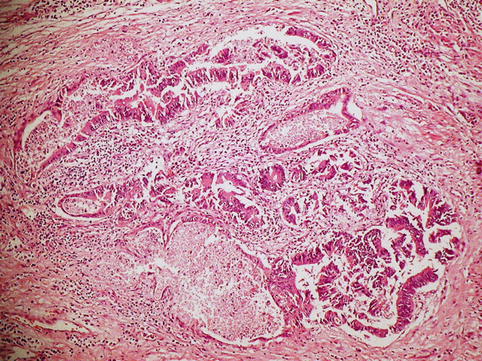
Fig. 11.2
Squamous cell carcinoma. The tumor consists of well-differentiated squamous cells forming keratin pearls (arrow)
Fig. 11.3
Adenocarcinoma. Note the glandular formation of the malignant cells (arrows)
There are tumors that do not follow any classification scheme, and they have been identified by trivial names, such as seminoma and melanoma. Other tumors carry eponyms, such as Hodgkin’s disease and Ewing’s sarcoma.
Tumors within a single organ or single type of epithelium are further subclassified into different types; each has its own characteristics, prognosis, and response to therapy. A comprehensive and detailed histological classification of tumors of various organ systems is presented in several reviews and monographs [6, 7]. Malignant tumors are neoplasms that extend into surrounding tissue without respecting normal tissue boundaries, are capable of invading lymphatics and blood vessels, and can be transported to distant sites. Several salient abnormalities are helpful to the pathologist in making the morphological diagnosis of such tumors, and they are expressed in two ways: first, there are abnormalities that affect individual cells in the form of cytological features and increased mitotic activity. Cytological features of malignancy include cell enlargement, increased ratio of nuclear to cytoplasmic area, pleomorphism (variation in size and shape), and chromatin clumping.
The tumor consists of well-differentiated squamous cells forming keratin pearls of nuclear chromatin and big nucleoli (Fig. 11.4). Second, there are abnormalities that affect intercellular relationship, i.e., altered orientation of neoplastic cells and stroma leading to disorganization [8].
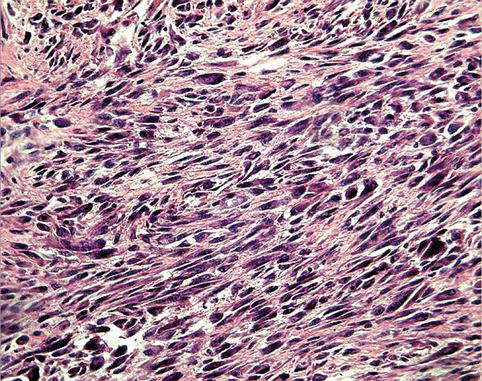
Fig. 11.4
Soft tissue sarcoma consisting of pleomorphic spindle cells, illustrating the features of malignancy as variable cell size and shapes and increased nuclear/cytoplasmic ratio
11.1.2 Grading
Grading is a scheme that attempts to determine the degree of malignancy and is based on the evaluation of certain parameters that vary according to the system used. These broadly include the following: degree of tumor cellularity, resemblance of tumor cells to their normal forbears morphologically and functionally, cellular pleomorphism or anaplasia, mitotic activity (number and abnormality), and necrosis [9]. In general, a three-grade system has proven to be the most reproducible; well, moderately, and poorly or undifferentiated, or grades I, II, and III, where grade III represents the least-differentiated tumor. Certain tumor types are defined as high grade, for example, rhabdomyosarcoma, Ewing’s sarcoma, and synovial sarcoma. Nonetheless, tumor grading has a few shortcomings: first, grading is subjective and depends on the pathologist’s judgement. Second, tumors are heterogeneous, and accurate grading requires microscopic examination of representative tissue. In these cases, the least-differentiated area of the tumor should determine the grade. Thus, the tumor grade is a qualitative assessment of the differentiation of the tumor expressed as the extent to which a tumor resembles the normal tissue at that site, expressed in numerical grades from most differentiated to least differentiated.
11.1.3 Tumor Staging
Staging of cancer depends on the size of the primary neoplasm, its extent to regional lymph nodes, and the presence or absence of metastasis. Cancer care is a cooperative, multidisciplinary endeavor; therefore, if the medical disciplines involved in cancer care are to work well together, they must be able to communicate with precision. The TNM system has been developed by the American (American Joint Committee on Cancer, AJCC) and European (Union International Contre le Cancer, UICC) commissions on cancer to allow systematic categorization and description of cancer patients. In this fashion, disease progression patterns natural history, and treatment outcome can be more reliably documented when applied to the individual patient.
The AJCC classification is based on the premise that cancers of similar histological type or site of origin share similar patterns of growth and extension. Obviously, cancers exhibit a variety of growth and extension patterns based upon unique features of either the host or the tumor. A classification scheme must therefore encompass all potential manifestations of both the tumor and its host. The objectives of a staging system can be briefly summarized as follows: (a) to aid the clinician in planning treatment, (b) to give some indication of prognosis, (c) to assist the evaluation of end results, (d) to facilitate the exchange of information between treatment centers, and (e) to assist in the continuing investigation of cancer. To meet the stated objectives, a system of classification is needed that has basic principles applicable to all anatomical sites regardless of treatment and in which clinical appraisal can be supplemented by later information from surgery, histopathology, and/or other technologies. The TNM system meets these requirements. It is an expression of the anatomical extent of disease and is based on the assessment of three components:
T – the extent of primary tumor
N – the absence or presence and extent of regional lymph node metastases
M – the absence or presence of distant metastases
Temporal variables are defined in the TNM classification scheme to allow an update to occur after certain data have been obtained: TNM clinical–diagnostic staging allows for pretreatment characterization via clinical examination and specific diagnostic studies. TNM surgical–evaluative staging is applied following a major surgical exploration or biopsy. TNM postsurgical treatment–pathological staging characterizes the extent of the cancer following thorough examination of the resected surgical specimen. TNM retreatment staging is applied in instances where the initial therapy has failed and additional treatment decisions are being considered. TNM autopsy staging is the final staging, done after the postmortem study.
11.1.4 Rate of Growth
Most benign neoplasms grow slowly, and most malignant neoplasms grow much faster. There are many exceptions to this generalization, however, and some benign neoplasms grow more rapidly than some cancers. For example, uterine leiomyomas (benign smooth muscle tumors) may increase rapidly in size during pregnancy (hormonal effect). The rate of growth of malignant tumors correlates in general with their level of differentiation. Rapidly growing malignant neoplasm often contains central areas of ischemic necrosis because the tumor blood supply, derived from the host, fails to keep pace with the oxygen needs of the expanding mass of cells [1].
11.2 Tumor Biology
Developing cancer cells must acquire a variety of characteristics not generally found in nontransformed cells. The cancer cell represents the culmination of a complex process of developing capacity for largely unregulated growth.
The main characteristics that differentiate the cancer cell from the precancerous or noncancerous cell are capabilities of self-sufficiency in growth signals, insensitivity to antigrowth signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and a potential for tissue invasion and metastasis. In addition, the enabling characteristic of genetic instability was noted as a driving force for acquiring these cell characteristics.
11.2.1 Cell Growth and Cell Cycle
Normal cell proliferation (also known as cell growth or cell division) is essential for tissue homeostasis in the adult body. When stimulated to divide, a normal cell progresses through a tightly regulated process known as the cell cycle.
This cycle is biochemically initiated by external stimuli, modulated by both external and internal growth controls. Certain genes and cell-cycle-specific proteins are activated and deactivated synchronously as the cell progresses through the different phases of the cell cycle. The cell cycle is a complex circuit composed of positive and negative protein regulators, the role of which is to duplicate DNA specifically and to segregate it evenly into two identical progenies.
The cell cycle has different phases. In G0 phase (gap 0 or resting phase), the cell is generally programmed to perform specialized functions. The other four distinct phases include G1 phase (gap 1 or interphase) in which proteins and RNA are synthesized for specialized cell functions. In late G1, a burst of RNA synthesis occurs, and many of the enzymes necessary for DNA synthesis are manufactured. The cellular contents excluding the chromosomes are duplicated. In S phase (DNA synthesis), the cellular content of DNA doubles, and each of the 46 chromosomes is duplicated.
During G2 phase (gap 2), DNA synthesis ceases and protein and RNA synthesis continues. The microtubular precursors of the mitotic spindle are produced. The cell “double-check” the duplicated chromosomes for any errors and making any needed repair.
In M phase (mitosis), the rates of protein and RNA synthesis diminish abruptly while the genetic material is segregated into daughter cells. After completion of mitosis, the new cells enter either the G0 or G1 phase (Fig. 11.3). When a cell leaves the dormant state of G0 and enters a metabolically active phase during G1, the destiny of the cell cycle pivots in the equilibrium as the decision to undergo division must be made at the restriction point [10]. Because G1 is such an essential phase of the cell cycle, it is not astonishing that many oncogenic perturbations have been found as targeted amplifications or mutations of G1-specific protein regulators.
Cyclins are special proteins that activate the various phases of the cell cycle. Cyclins combine with, activate, and direct the action of special tyrosine kinases called cyclin-dependent kinases. Cyclins specific for various phases of the cell cycle rise and fall in synchrony with the progression of the cell through the cell cycle.
Normal cells have mechanisms or checkpoints that detect abnormalities in DNA sequences. When DNA is damaged, a number of repair mechanisms replace damaged nucleotides with normal molecules. These mechanisms ensure that new genetic material in daughter cells is an exact copy of that of the parent cell. The first checkpoint occurs toward the end of G1 phase, just before cells enter the S phase. If injuries are detected, they either are repaired or the cell is made to undergo apoptosis.
The second checkpoint occurs just before the cell enters M phase. The cell-cycle inhibitors stop the cell until it is determined whether the new progeny has the genetic copies of the parent. Mitosis will not begin if the cell has not completely and accurately replicated its entire DNA. This process is mediated by decreased activity of cyclin-associated kinases and tumor suppressor proteins.
Although some cell types are constantly dividing to replenish tissue such as cells in the basal layer of skin, many cells exist in a nonproliferative state (G0) or quiescence. Normal cells are continually regulated by antigrowth signals that maintain them either in a quiescent state (waiting to reenter the cell cycle if needed) or in a permanent nonproliferative state. Quiescent cells typically proliferate only in response to appropriate mitogenic growth signals.
Normal tissue maintains a stable cell population through two distinct growth and antigrowth signaling networks that regulate the cell-cycle machinery. Signaling in both networks begins when extracellular molecules called as ligands bind transmembrane receptors, resulting in the propagation of a signal across the cell membrane to downstream intracellular molecules that receive and transmit the signal through the cytoplasm into the nucleus. As mentioned before, this process is known as signal transduction pathway. At the nucleus, either a growth signal leads to the activation of nuclear transcription factors that regulate the gene expression leading to cell-cycle progression or an antigrowth signal blocking these molecules.
Extrinsic growth or antigrowth stimuli can come from several sources outside of the cell including soluble growth factors, extracellular matrix components, or other cell-surface molecules. Cancer cells can achieve growth autonomy by internally producing abnormal amounts of their own mitogenic growth factors. This is known as autocrine stimulation. Also, cancer cells may stimulate the neighboring cells in the tumor microenvironment. Mitogen expression by neighboring cells is known as paracrine stimulation antigrowth signaling.
Although overexpression of growth factors is frequently involved in cancer cell proliferation, less is known about the role of antigrowth factors in the evasion of antiproliferative signals. Transforming growth factor-beta (TGF-β) is an antigrowth factor expressed by most normal epithelial cells. TGF-β functions in an autocrine fashion to inhibit proliferation and therefore may be a tumor suppressor protein. Recent evidence suggests that the loss of endogenous TGF-β expression may provide a growth advantage to cancer cells. Cancer cells can also turn off expression of TGF-β receptors, making them less responsive to exogenous TGF-β.
Antigrowth signaling can be disabled or disrupted through mutations in transmembrane receptors. The mutations lead to the expression of fewer receptors or dysfunctional receptors that are less responsive to normal levels of extracellular antigrowth signals. The best known example is the TGF-β receptor. Also, insensitivity to antigrowth signals can occur when intracellular signaling pathways are disrupted and become unresponsive to TGF-β signaling or other antigrowth stimulation [11, 12].
11.2.2 Tumor Neovascularization (Angiogenesis)
Cell survival and proliferation is dependent on an adequate supply of oxygen and nutrients and the removal of toxic metabolites. Tumor cells still require nutrients and oxygen in order to grow. Since oxygen can diffuse radially from capillaries for only 150–200 μm, the growth of tumor masses greater than 1 mm in diameter depends on the formation of new blood vessels, also known as tumor neovascularization or angiogenesis.
Neovascularization is a feature of neoplasia and is initiated, maintained, and controlled by multiple molecules that are released from tumor cells, endothelial cells, and other cell types. Neovascularization supplies nutrients and oxygen. Additionally, endothelial cells of the vessels stimulate the growth of adjacent tumor cells by secreting different factors, such as insulin-like growth factor, platelet-derived growth factor (PDGF), granulocyte–macrophage colony-stimulating factor (GM-CSF), and interleukin (IL)-1. Angiogenesis is required not only for continued tumor growth but also for metastasis [1].
The new tumor vessels are formed from endothelial cells of the blood vessels within the tumor site. Additionally, circulating endothelial cell progenitors may home to sites of ongoing angiogenesis, where they differentiate into mature endothelial cells and contribute to angiogenesis.
Tumor-associated angiogenic factors may be produced by tumor cells or may be derived from inflammatory cells (e.g., macrophages) that infiltrate tumors. The two most important tumor-associated angiogenic factors are vascular endothelial growth factor (VEGF) and basic fibroblast growth factor. The tumor cells not only produce angiogenic factors but also induce antiangiogenesis molecules. The molecular basis of the angiogenesis is not entirely clear but may involve increased production of angiogenic factors or loss of angiogenesis inhibitors. Hypoxia within the growing tumor favors angiogenesis by release of hypoxia-inducible factor-1 (HIF-1) [1]. Under normoxic conditions (20 % oxygen), HIF is rapidly degraded through the von Hippel–Lindau tumor suppressor protein. Under hypoxic conditions, HIF target genes are transcribed then activated. HIF-1 controls transcription of VEGF. The transcription of VEGF also is under the control of RAS oncogene, and RAS activation upregulates the production of VEGF. The wild-type TP53 gene seems to inhibit angiogenesis by inducing the synthesis of the antiangiogenic molecule thrombospondin-1. With mutational inactivation of both TP53 alleles (a common event in many cancers), the levels of thrombospondin-1 drop precipitously, tilting the balance in favor of angiogenic factors [13].
Increased tumor vascularization and increased levels of angiogenic inducers are associated with increased risk of tumor metastases and shortened survival in patients with cancer [14]. Angiogenic inhibitors found naturally in the body (endogenous inhibitors) act as negative regulators of endothelial cell proliferation and migration. Natural angiogenic inhibitors include thrombospondin, angiostatin, endostatin, and others [15].
Because of the important role of angiogenesis in tumor growth, extensive publication concentrated on antiangiogenesis therapy and the results are very promising [16–23]. Therapeutics targeting angiogenesis have already been used successfully for the treatment of breast, lung, kidney, and colon cancers. Several types of antiangiogenic drugs have been developed including agents that target intracellular signals within endothelial cells and tumor cells, agents that block external angiogenic stimuli, and agents that inhibit the breakdown of the extracellular matrix.
The VEGF pathway has also been targeted by antiangiogenic drugs which inhibit the tyrosine kinase receptors of several angiogenic factors, through targeting the cytoplasmic portion of the receptor. These drugs block cell division and inhibit new blood vessel growth. A major advantage of antiangiogenic therapy is that it targets a cell population (endothelial cells) with relative genetic stability compared with other cancer cells [15, 24, 25].
11.2.3 Distinguishing Features of Tumor Cells
Cancer is a genetic disease resulting from multiple, sequential genetic changes affecting oncogenes, tumor suppressor genes, and modifiers [10, 26–29]. Because of this multistep process, most human malignancies show various degrees of genetic heterogeneity even if they originate from single cells.
The multistep progression model determines that cells pass through a number of distinctive intermediate stages of evolution from normalcy to full malignancy [30]. The evolved tumor cells vary significantly from their normal counterparts.
Tumor cells have distinguishing morphological and structural features that are different from those of the cells of origin. Moreover, the abnormal cells show altered interaction with neighboring cells.
11.2.3.1 Loss of Contact Inhibition of Growth
Normal cells have an ordered growth pattern and a predicted relationship with their neighboring cells, and that growth pattern is predominantly two dimensional. Further normal cell division is inhibited by contacts made with other cells; this is the phenomenon of contact inhibition [23]. In contrast, tumor cells exhibit loss of contact inhibition and continue to display a disordered growth pattern.
11.2.3.2 Growth Regulatory Response Pattern
Another feature that distinguishes normal growth from malignant proliferation is the reduced dependence of the latter on the presence of the known stimulatory and inhibitory growth factors. The diversion from the control of the growth regulatory factors can be explained, in part, by the discovery of biochemical changes within cancer cells resulting from some genetic alterations. Thus, when normal cells are grown in culture, they continue to divide for a limited number of generations and then experience a senescent crisis, in which most cells stop dividing and die, and no cells survive to establish permanent cell lines. On the other hand, human tumor cells usually have an unlimited potential for growth and are thus immortalized [31, 32].
This distinctive growth factor independence or autonomy has been attributed to at least four different mechanisms. First, tumor cells have the ability to secrete mitogenic growth factors that have a growth-stimulatory ability on the same cell that has released them, resulting in an autocrine positive-feedback loop [33]. Second, normal cells display growth factor receptors on their surfaces. These receptors release growth-stimulatory signals into the cell when they bind their cognate ligands. Alteration in number or structure can result in the release of mitogenic signals into the cell, even in the absence of any growth factor signals [34]. Therefore, these aberrantly expressed or structured receptors then operate as oncogene proteins. The best example of this mechanism is illustrated by the overexpression of HER-2/neu proto-oncogene in mammary carcinoma [35]. Third, there is a cytoplasmic signaling pathway responsible for picking up signals from cell-surface receptors and transducing them to central growth regulatory switches within the cell. It appears that proteins of such genes as RAS participate in signal-transducing events in these pathways [36]. Finally, the growth factor independence of tumor cells may also be attributed to the behavior of nuclear proto-oncogenes that are normally regulated through a wide range of expression [35, 36].
11.2.3.3 Immune Evasion
The immune system consists of many specialized cell types that collectively protect the body from bacterial, parasitic, fungal, and viral infections. The immune system is also thought to play a role in the detection and elimination of malignant cells.
Immune cells can identify cancer cells that express tumor-specific antigens (molecules unique to cancer cells) or tumor-associated antigens (molecules differentially expressed by cancer cells and normal cells). Antigens are generated by proteasomes that break down intracellular proteins into short peptides. These peptides are presented on the cell surface in major histocompatibility complex molecules.
Another characteristic feature of human malignancy is its ability to escape the human immune surveillance pathways. It is known that human tumor cells express on their surfaces novel antigens that are not present on the surfaces of their untransformed progenitors. One type of novel antigen may be common to many different types of malignancy and may be recognized by the natural killer lymphocytes even without specific prior immunization [37]. In other instances, novel antigens specific to a particular type of tumor may be displayed. According to one theory of tumorigenesis, all individuals develop abundant transformed cells over the course of their lives, but most of these cells are recognized and destroyed by one or another module of the host’s immune mechanisms. Support for this theory stems from the observation of the substantial increase in the incidence of malignancy in patients with acquired immune deficiency syndrome, in patients with organ transplants receiving intensive immunosuppressive agents, and in patients with other immune deficiency disorders. It has been proven that tumor cells may escape the host immune system by downregulating the expression of HLA antigens, which normally assist lymphocyte recognition of the target cells.
Cancer cells can efficiently block the immune system in several ways. Metastatic growth requires cancer cells to acquire the capability of eliciting immunosuppression. Tumors can induce immunosuppression through a variety of mechanisms such as expression of immunosuppressive molecules and induction apoptosis in lymphoid cells.
An example of an immunosuppressive molecule is the transforming growth factor-β (TGF-β). The TGF-β pathway is utilized by regulatory T cells to suppress immune overreactions and avoid autoimmunity. However, tumors can also overexpress TGF-β to suppress cytotoxic T-cell function and thereby escape immune control. Additionally, regulatory T cells can invade the tumor microenvironment, where they produce ample amounts of TGF-β that suppress antitumor immune responses [38].
A relatively new approach to cancer therapy is the use of vaccines to boost antibody production or elicit T-cell responses against cancer cells. Therapeutic cancer vaccines are designed to expose the immune system to a tumor antigen using a range of strategies.
11.2.3.4 Metabolic Alterations
Tumor cells exhibit a vast array of metabolic differences distinguishing them from their untransformed counterparts. This is illustrated primarily in simplified metabolic activities and by an increased synthesis of material necessary for cell division. Some of the most striking metabolic alterations include the utilization of anaerobic pathways and the increased utilization of glucose transport [39].
11.2.4 Invasion and Metastasis
Metastasis is a multistep process in which tumor cells invade nearby tissues and colonize other parts of the body. Malignant neoplasms disseminate by one of three pathways: (1) seeding within body cavities (an example of this is carcinoma of the colon that may penetrate the wall of the gut and reimplant at distant sites in the peritoneal cavity), (2) lymphatic spread which is the preferred way of spread by carcinomas in general, or (3) hematogenous spread which is favored by sarcomas. A complex set of biological functions must be acquired for a primary tumor to progress to metastatic disease. The ability to metastasize is a multifaceted phenomenon which requires several prerequisites [40]: (a) local invasion (invasion by tumor cells through adjacent structures), (b) intravasation (entrance of tumor cells into blood or lymphatic vessels), (c) survival of tumor cells within the circulation and avoidance of the immune system, (d) extravasation, and (e) colonization (implantation in a foreign tissue with establishment of a new tumor locus).
Most carcinomas arise in epithelial cell layers with an underlying basement membrane. Invasive tumors frequently secrete enzymes, including collagenases, heparanase, and stromelysin, that are capable of degrading this type of physical barrier [41]. Once a tumor has eroded through the wall of a blood or lymphatic vessel, individual tumor cells may detach and circulate through the body as an embolus. Encasing these cells in cots of fibrin or in aggregates of platelets may protect them from destruction by the immune system. The presence of tumor cells in the circulation does not necessary lead to metastases [42]. However, these circulating cells have to “home” preferentialy to a specific target organ. This pattern has been described as the “seeds” and “soil” model and has been demonstrated experimentally and observed consistently in oncological practice. It is not clear how these patterns of metastases arise, but some evidence suggests that tumor cells can respond to specific chemotactic signals. Moreover, specific receptors have recently been identified on the surfaces of metastasizing tumor cells that cause them to adhere to complementary structures displayed by endothelial cells in certain organs [43].
As mentioned earlier, the metastatic process begins with the dissociation of cancer cells from adjacent stromal cells and the extracellular matrix, followed by proteolytic degradation of the extracellular matrix and migration toward lymphatic or blood vessels, invasion through the vessel wall, and transportation through the circulation. Cancer cells may also gain direct entry into the circulation through new vessel growth within the tumor (angiogenesis or lymphangiogenesis).
Although cancer cells can remain and grow within the intravascular space, they often migrate out of the vascular space and into a target organ. The arrest of circulating cancer cells at distant sites is thought to occur through size-dependent trapping or adhesive interactions with the vessel wall.
Metastatic cancer cells might progress or remain dormant micrometastases for months to decades within the target organ site. Successful colonization into macrometastases occurs only after adaptation to the new microenvironment and may require additional mutations in the cancer cell population, adjacent stromal cells, or both.
Invasion of cancer cells into the surrounding stroma is prevented by cell–cell and cell–matrix adhesion molecules. Loss of these adhesive attachments enables cancer cells to detach from the primary tumor site. Cell adhesion molecules are grouped into four distinct families: cadherins (cell–cell), integrins (cell–matrix), immunoglobulins (cell–cell), and selectins (cell–cell).
Both intravasation and extravasation describe the process of transendothelial migration of cancer cells through the blood vessel wall. Many signaling molecules secreted by cancer cells cause disruption of the vessel wall, creating openings between endothelial cells and penetration, thereby gaining access of cancer cells to the vessel lumen. Only a small fraction of metastatic cells survive during transport through the circulation, and those that do must surmount a variety of natural barriers. Cancer cells with mutations are capable of continued proliferation.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


