CANCER BIOLOGY Prerit Sharma, Abhishek Mahajan, Shreya Shukla, Shivam Rastogi, Pritesh Shah Cancer is a multifactorial disease, and imaging forms an important platform to demonstrate various mechanisms and phases of pathogenesis. Combining multiple imaging modalities and biomarkers improves diagnostic sensitivity and specificity. As a result, biomedical imaging plays a significant role in cancer management. These include prediction, screening, biopsy guidance for detection, staging, prognosis, therapy planning, therapy guidance, therapy response, recurrence and palliation. The basis of modern pathology is understanding the cellular and molecular aberrations that give rise to diseases. This chapter discusses basic principles and emphasizes recent advances relevant to cancer biology (Table 2.15.1.1). Reproduced from Vinay Kumar, Abul Abbas, Jon Aster. Robbins Basic Pathology, 10e, The Cell as a Unit of Health and Disease, Table 1.1, Philadelphia, Elsevier Inc., 2018. Approximately 3.2 billion D.N.A. base pairs constitute the human genome. Of this entire genome, there are only about 20,000 protein-encoding genes (1.5%). These serve as the blueprints that instruct the assembly of enzymes, structural elements, and signalling molecules for an approx. The number of 50 trillion cells together constitutes the physical body. There are five major classes of available non–protein-coding sequences in the human genome (Fig. 2.15.1.1): Eukaryotic cells are different from bacterial or archaeal cells as they contain a membrane-bound nucleus. In addition, highly organized physical structures called intracellular organelles such as the endoplasmic reticulum, Golgi apparatus, chloroplasts and mitochondria are found in eukaryotic cells. The physical and chemical constituents of the cell organelles hold great significance for normal cell functioning. The cell membrane (a.k.a. plasma membrane) is a thin, pliable, elastic structure surrounding the eukaryotic cell. It is about 7.5–10 nm in thickness and is composed almost entirely of proteins and lipids. It regulates the passage of organic molecules, ions, water, and oxygen into and out of the cell. In addition, waste products (such as CO2 and ammonia) leave the cell by passing through the cell membrane, often requiring the assistance of protein transporters. One of the essential functions of the plasma membrane is protection and nutrient acquisition. This function is accomplished with the help of membrane components such as phosphatidylinositol, phosphatidylserine, glycolipids and sphingomyelin. The plasma membrane is engraved with a variety of proteins and glycoproteins, whose functions are namely: Many proteins that constitute the plasma membrane function as large complexes. Most proteins are integral or transmembrane proteins. These have one or more hydrophobic α-helical segments that traverse the lipid bilayer. Post-translational modification can be done on proteins synthesized on free ribosomes within the cytosol. The methods include adding prenyl groups (e.g., farnesyl, associated with cholesterol) or fatty acids (e.g., palmitic or myristic acid) that insert into the cytosolic side of the cell wall. Post-translationally added glycosylphosphatidylinositol tails anchor proteins on the extracellular face of the membrane. Peripheral membrane proteins may bond with true transmembrane proteins by covalent bonds. The transport of molecules across the lipid bilayer or into the intracellular sites via vesicular transport occurs via several mechanisms such as (1) Passive Diffusion; (2) Carriers and Channels; and (3) Receptor-Mediated and Fluid-Phase Uptake. An intracellular scaffold of structural proteins from the cytoskeleton determines cells’ ability to adopt a specific shape, maintain polarity, organize intracellular organelles and migrate. The eukaryotic cells have three major classes of cytoskeletal proteins: The junctional complexes form mechanical links between cells and facilitate receptor-ligand interactions. The cells connect and communicate via these junctional complexes. Similar junctional complexes also mediate interaction with the E.C.M. Functionally, the most significant of these junctions are Both minuscule and large dispersed particles and organelles are dispersed within the cytoplasm. Cytosol is the jelly-like component of the cytoplasm which is made up of dissolved electrolytes, glucose and proteins. Dispersed within the cytoplasm are neutral fat globules, glycogen containing granules, ribosomes, secretory vesicles and five vital organelles: the endoplasmic reticulum, the Golgi body, mitochondria, lysosomes and peroxisomes. Biosynthetic Machinery: Endoplasmic Reticulum and Golgi Apparatus In an ongoing process of balancing synthesis and degradation, all cellular constituents, including structural proteins, enzymes, transcription factors and even the phospholipid membranes, are constantly renewed. Endoplasmic Reticulum (E.R.) is a network of flat, tubular, vesicular structures in the cytoplasm. This organelle aids in processing molecules produced by the cell and transports these molecules to their destinations inside or outside the cell. Their walls are composed of lipid bilayer membranes containing large proteins, like the cell wall. Ribosomes are minute granular particles attached to the endoplasmic reticulum’s outer surfaces. The reticulum is named the granular endoplasmic reticulum, where these particles are present. The ribosomes are composed of a mix of R.N.A. and proteins. Their primary function is to synthesize new protein molecules. Part of the endoplasmic reticulum that has no attached ribosomes is called the agranular or smooth endoplasmic reticulum. Smooth E.R. synthesizes lipid substances and is responsible for other processes of the cells promoted by intra-reticular enzymes. Golgi apparatus has membranes similar to the agranular endoplasmic reticulum. It functions in close relation to the endoplasmic reticulum and is composed of four or more stacked layers of thin, flat, enclosed vesicles lying in proximity of the nucleus. This apparatus is present in abundance in secretory cells on the side from where the secretory substances are extruded. Waste Disposal: Lysosomes and Proteasomes The cells depend primarily on lysosomes to degrade internalized material and accumulated internal waste. There are, however, multiple other routes to degrade intracellular macromolecules. Lysosomes: They are vesicular organelles formed by the disintegration of the Golgi body and subsequent dispersion in the cytoplasm. The lysosomes permit the cell to digest: Hydrolytic enzymes are highly concentrated in lysosomes which function to split the organic substances into smaller substances like amino acids and glucose which facilitates higher rate of diffusion. Peroxisomes: Similar in structure to lysosomes are peroxisomes. Although lysosomes and peroxisomes differ in two critical ways, peroxiomes are formed by self-replication or budding from the smooth endoplasmic reticulum instead of the Golgi body. Second, they contain oxidases rather than hydrolases. Many oxidases can form hydrogen peroxide (H2O2) by combining oxygen with hydrogen ions derived from different intracellular chemicals. Cellular Metabolism and Mitochondrial Function The mitochondria are known as the ‘powerhouses’ of the cell. Without them, cells will not be able to extract enough energy from the nutrients. Consequently, all cellular functions would cease. It comprises two lipid bilayers: an outer membrane and an inner membrane. The infoldings of the inner membrane form shelves onto which oxidative enzymes are attached. These shelves are called cristae and serve as a platform for chemical reactions. The matrix forms the inner cavity of the mitochondrion, which contains large quantities of dissolved enzymes that are essential for deriving energy from nutrients. These enzymes cause oxidation of the nutrients by operating in association with the oxidative enzymes on the cristae. Carbon dioxide and water are the by-products. There is the simultaneous release of energy. The released energy synthesizes a ‘high-energy’ substance called adenosine triphosphate (ATP). ATP is then carried out of the mitochondrion, where it diffuses throughout the cell to release its energy for performing cellular functions. The nucleus is the cell’s control centre. It sends messages to the cell to grow and mature participate in cell replication, or die. It contains large quantities of D.N.A., which together form the genes are responsible for the various cell protein characteristics. These cell proteins include both the structural proteins and the intracellular enzymes that in turn control the cytoplasmic and nuclear activities. Another important function of the genes is to control and promote the reproduction of the cell by initial replication to make two identical sets of genes. Every cell duplicates by mitosis to produce two daughter cells. Each daughter cell receives one set of D.N.A. genes. The nuclear membrane comprises two separate layers namely inner and outer. The outer nuclear membrane is contiguous with the membranes of the endoplasmic reticulum while the space between the two nuclear membranes continues into the space inside the endoplasmic reticulum. Cellular Activation Cell Signalling: The signals that most cells respond to can be classified into the following groups: Danger and pathogens, cell–cell contacts mediated through adhesion molecules and/or gap junctions, cell-ECM contacts mediated through integrins, secreted molecules. Signalling pathways can also be classified based on the spatial relationships between the sending and receiving cells: Paracrine signalling, autocrine signalling, synaptic signalling, endocrine signalling. All extracellular stimuli (i.e., paracrine, autocrine, synaptic, or endocrine) convey their signal to the cell via specific receptor proteins. Receptors may be present on the cell surface, generally transmembrane proteins with extracellular domains that bind activating ligands. In addition, receptors may be present within the cell, termed intracellular receptors, including transcription factors activated by lipid-soluble ligands that can easily transit plasma membranes. Vitamin D and steroid hormones that activate nuclear hormone receptors are good examples. Signal Transduction Pathways: The interaction of a cell-surface receptor and its ligand activates signalling through ligand-induced clustering of the receptor (receptor cross-linking) or by inducing a physical change in receptor structure. These mechanisms result in a conformational change in the receptor’s cytosolic tail, resulting in additional intracellular biochemical events. Modular Signalling Proteins, Hubs and Nodes: Any initial signal that relies on enzymatic activity impacts multiple processes, each is contributing to the outcome, with adapter proteins playing a pivotal role in organizing intracellular signalling pathways. For example, specific phosphorylation of any given protein allows it to associate with a host of other molecules, thereby causing various processes such as enzyme activation (or inactivation), nuclear (or cytoplasmic) localization of transcription factors, activation or inactivation of transcription factor, polymerization (or depolymerization) of actin, degradation or stabilisation of proteins, activation of feedback inhibitory or stimulatory loops. Transcription Factors: Most signal transduction pathways ultimately induce long-lasting effects on cellular function by modulating gene transcription; this occurs through the activation and/or nuclear localization of transcription factors. Transcription factors often contain: Interactions mediated by these domains can be controlled by posttranslational modifications such as phosphorylation. These changes can result in translocation from the cytoplasm into the nucleus, modify transcription factor protein half-life, expose specific D.N.A. binding motifs, or promote binding to components of the R.N.A. polymerase complex to augment transcription factor activity. Growth Factors and Receptors Growth factors stimulate signalling pathways and genes that augment cell survival, growth, and division. Binding of the growth factors to specific receptors ultimately influences the expression of genes leading to various processes such as: The E.C.M. comprises a protein network that constitutes a large proportion of any tissue. Therefore, cell interactions with the E.C.M. are critical for developing, healing and maintaining standard tissue architecture. Apart from the primary function of ‘space-filling’ around the cells, the E.C.M. also serves as a: The E.C.M. occurs in two primary forms: E.C.M. components can be further categorized as (Fig. 2.15.1.3): Cell proliferation plays a primary role in organism development for maintaining steady-state tissue homeostasis and replacing dead or damaged cells. It comprises of the following two consecutive processes: Initially, the cell division process was studied into two subheadings: Cells in G1 can also enter a resting state called the G0 phase which accounts for the non-growing, non-proliferating cells in the human. Each stage requires completing the previous step and subsequent activation of necessary factors. Non-fidelity D.N.A. replication or cofactor deficiency leads to arrest at different transition points. Cyclin and Cyclin-dependent kinases (CDKs): The cell cycle is regulated by the activators and inhibitors. Cell cycle is supervised mainly by: Cyclins are found in every cell throughout all eukaryotes. The cyclin family is quite largewith 30 identified members to date (N.H. Lents, J.J. Baldassare, in Encyclopedia of Cell Biology, 2016). Different cyclins function in different cell cycle phases along with their associated CDK, and different subtypes show specific tissue-distribution patterns in multicellular organisms. The cyclins D, E, A and B appear sequentially during the cell cycle and bind to at least one or more CDKs. The cell cycle thus resembles a relay during which each leg is regulated by a definite set of cyclins with a different set of cyclins taking charge one after the other. In addition, surveillance mechanisms primed to sense D.N.A. or chromosomal damage are embedded within the cell cycle. These quality control checkpoints ensure that cells with genetic aberrations cannot complete replication. Thus, the G1-S checkpoint monitors D.N.A. integrity before irreversibly committing cellular resources to D.N.A. replication. Later on, during the cell cycle, the G2-M restriction point ensures that there has been precise genetic replication before the cell divides. When cells detect D.N.A. irregularities, checkpoint activation delays cell cycle progression and triggers D.N.A. repair mechanisms. If the genetic abnormality is severe enough not to be repaired, cells either undergo apoptosis or enter a non-replicative state called senescence primarily through p53-dependent mechanisms. Transiently increased synthesis of a particular cyclin thus results in increased kinase activity of the CDK binding partner. As the CDK completes its round of phosphorylation, the associated cyclin is degraded, and CDK activity stops. Consequently, as cyclin levels rise and fall, the activity of associated CDKs will likewise increase and decrease (Fig. 2.15.1.4). CDK inhibitors (CDKIs) are responsible for enforcing the cell cycle checkpoints by modulation of CDK-cyclin complex activity. There are several different CDKIs, namely, In cancer, unrestrained cell proliferation is achieved by fundamental alterations in the genetic control of cell division by mutations occurring either in protooncogenes and/or in tumour suppressor genes. In normal cells, the products of protooncogenes act at diverse levels along the pathways that stimulate cell proliferation. Mutated forms of protooncogenes or oncogenes can promote tumour growth. Inactivation of tumour suppressor genes like p53 and pRb leads to the dysfunction of proteins that generally inhibit cell cycle progression. Cell cycle deregulation with cancer occurs through mutation of proteins essential at various levels of the cell cycle. Cancers are associated with mutations in genes encoding CDK, cyclins, CDK-activating enzymes, C.K.I., CDK substrates and checkpoint proteins. Many chemotherapeutic therapies use drugs presently either in clinical use or under development, target G1 CDKs, other cell cycle kinases (A.T.M. and A.T.R., CHK1 and CHK2, WEE1, Auroras, polos, MPS1), D.N.A. damage sensors (PARP), kinesins and epigenetic regulators (SETD2 methylase). They are being combined with one another or with conventional cytotoxic agents to advance cancer therapy (Fig. 2.15.1.5). Stem cells possess self-renewing properties and further differentiate into specific cells and tissues. Although some researchers separate stem cells into multiple subsets, there are fundamentally only two varieties: embryonic stem cells and tissue stem cells. The burgeoning field of regenerative medicine is based on identifying, isolating, expanding and transplant stem cells. It should, however, be remembered that this summary is intentionally brief, and one can readily find detailed information about topics reviewed here in textbooks and online literature devoted to cell and molecular biology. Cancer is one of the leading causes of death in India. While globally, non-communicable diseases (NCDs) accounted for 71% of total deaths, NCDs were estimated to account for 63% of all deaths, with cancer contributing to around 9% of total deaths in India. The projected number of patients with cancer in India was 1,392,179 for 2020. The five leading cancer sites are breast, lung, mouth, cervix uteri, and tongue (Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018 Nov.; 68(6):394–424.) make a reference out of this in the end. Even more excruciating than the mortality associated with cancers is the emotional and physical trauma inflicted. Some cancers, like Hodgkin lymphoma, are curable, whereas others, like pancreatic adenocarcinoma, are virtually always fatal. The only hope for controlling cancer lies in learning more about its causes and pathogenesis. Fortunately, great strides are constantly being made in understanding its molecular basis. Some excellent study results have emerged: cancer mortality for both men and women in the United States has declined during the last decade of the 20th century. It has continued its downhill course in the 21st century. The use of routine radiographic imaging provides a promising platform for linking specific imaging traits with respective gene expression patterns, further highlighting the underlying cellular pathophysiology. The imaging features assist molecular surrogates that contribute to the diagnosis, prognosis, and gene-expression-associated treatment response associated with diverse forms of human cancer. Gene expression profiling relies on the surgical procurement of tissue, involving several risks and potential complications and making it an impractical option for every cancer patient. Radiographic imaging studies in contrast are part of routine clinical care. It is mainly used to gather important anatomical and morphological information and is not perceived to imply much consequential molecular detail. Methods that assist in analyzing lesion characteristics such as permeability and blood flow, like dynamic contrast-enhanced (D.C.E.) magnetic resonance imaging (M.R.I.) or dynamic computerized tomography, are equipped to distinguish benign from malignant lesions. These can define the topographical boundaries of the cancer lesion, thereby improving staging accuracy. Neoplasia means ‘new growth’ in Greek, and thereby, a collection of cells and stroma composing new growths is termed a neoplasm. The tumour was described initially as swelling caused by inflammation but is now equated with neoplasm. Oncology (Greek oncos = tumour) is defined as the study of tumours or neoplasms. In the modern era, a neoplasm is defined as a genetic disorder of cell growth triggered by acquired or less commonly inherited mutations affecting a single cell and its clonal progeny. All tumours are composed of following main components: Connective tissue and derivatives Fibroma Lipoma Chondroma Osteoma Fibrosarcoma Liposarcoma Chondrosarcoma Osteogenic sarcoma Blood vessels Hemangioma Angiosarcoma Lymph vessels Lymphangioma Lymphangiosarcoma Mesothelium Benign fibrous tumor Mesothelioma Brain coverings Meningioma Invasive meningioma Blood Cells and Related Cells Hematopoietic cells Leukemias Lymphoid tissue Lymphomas Muscle Smooth Leiomyoma Leiomyosarcoma Striated Rhabdomyoma Rhabdomyosarcoma Stratified squamous Squamous cell papilloma Squamous cell carcinoma Basal cells of skin or adnexa Basal cell carcinoma Epithelial lining of glands or ducts Adenoma Papilloma Cystadenoma Adenocarcinoma Papillary carcinomas Cystadenocarcinoma Respiratory passages Bronchial adenoma Bronchogenic carcinoma Renal epithelium Renal tubular adenoma Renal cell carcinoma Liver cells Hepatic adenoma Hepatocellular carcinoma Urinary tract epithelium (transitional) Transitional cell papilloma Transitional cell carcinoma Placental epithelium Hydatidiform mole Choriocarcinoma Testicular epithelium (germ cells) Seminoma Embryonal carcinoma Tumors of Melanocytes Nevus Malignant melanoma Salivary glands Pleomorphic adenoma (mixed tumor of salivary origin) Malignant mixed tumor of salivary gland origin Renal anlage Wilms tumor Totipotential cells in gonads or in embryonic rests Mature teratoma, dermoid cyst Immature teratoma, teratocarcinoma Reproduced from Vinay Kumar, Abul Abbas, Jon Aster. Robbins & Cotran Pathologic Basis of Disease: South Asia Edition, Neoplasia, Table 7.1, New Delhi, Elsevier India, 2014. Non-lethal genetic damage is the major cause of carcinogenesis. A tumour is formed due to clonal expansion of one precursor cell that has incurred genetic damage (i.e., tumours are clonal). The principal targets of cancer-causing mutations are four classes of genes that regulate programmed necrosis (apoptosis). These genes are liable for D.N.A. repair, growth-promoting protooncogenes and growth-inhibiting tumour suppressor genes. Carcinogenesis results from the cumulation of mutations over time. Once established, tumours evolve genetically during their growth and progression under the pressure of Darwinian selection (survival of the fittest). As predicted, these studies identified two kinds of mutations were identified: (1) mutations that are present altogether tumour sites tested, which were presumably present in the cell of origin at the instance of transformation and (2) mutations that are unique to a subset of tumour sites, which were likely acquired after transformation during the outgrowth and spread of the tumour. The natural transformation of cancer and changes in tumour behaviour following therapy abide by the theory of the selection of the fittest cells. In addition to D.N.A. mutations, epigenetic aberrations also contribute to the malignant properties of cancer cells. Epigenetic modifications include D.N.A. methylation, which silence gene expression, and modifications of histones, the proteins that package D.N.A. into chromatin, which may enhance or dampen genetic expressivity. Self-Sufficiency in Growth Signals Oncogenes are mutated genes that cause excessive cell growth, even without growth factors and other growth-promoting external cues. A significant discovery in cancer was that oncogenes are mutated or overexpressed versions. Physiologic growth factor-induced signalling is studied mainly under following headlines: Aberrations in multiple signalling pathways have been observed to play a significant role in the formation of neoplasms. Components of these pathways serve as oncoproteins when mutated. Whereas normal cells regulate cell growth and division to maintain cell homeostasis, cancer cells can frequently proliferate. The assembly of protein ligands accomplishes proliferation. These send signals to normal cells to supply growth factors, increasing receptor proteins on the neoplastic cell surface to make them sensitive to growth factor ligands. Epidermal growth factor receptor (EGFR) is a trans-membrane glycoprotein with two main components, an extracellular epidermal growth factor binding domain and an intracellular tyrosine kinase domain that controls signaling pathways regulating cellular proliferation. Upon binding of a specific ligand, there is conformational change in the normally functioning EGFR and phosphorylation of the intracellular domain occurs, causing downstream signal transduction by various pathways such as the Raf1-extracellular signal-regulated kinase, PI3K/Akt, and signal transducer and activator of transcription (STAT) factors. Depending on the pathway, there is inhibition of apoptosis withthe common end result being cell proliferation or cell maintenance. Overexpression of EGFR or mutations in intracellular EGFR has been observed in 43%–89% cases of non-small cell lung cancer (Bethune G et al, Journal of thoracic disease, 2010). c-Kit, a receptor tyrosine kinase, is involved in intracellular signalling. The mutated form of c-Kit plays a significant role in the occurrence of some cancers, leading to the concept that inhibiting c-Kit kinase activity can be a target for cancer therapy (Babaei MA, Drug design, development and therapy, 2016). After ligand binding, K.I.T. causes phosphorylation and subsequent activation of downstream members of the Ras-Raf-MAPK, Jak/Stat, and PI3k/A.K.T. signalling pathways, thus, controlling cell proliferation, apoptosis, chemotaxis, and metabolism. Gain of function mutations in K.I.T. is present in 55% of gastrointestinal stromal tumours (GISTs). EGFR may be a tyrosine kinase receptor comprising four receptors: ErbB1 (EGFR/HER1), ErbB2, ErbB3, and ErbB4 (HER4). E.G.F.R. is overexpressed in several human tumours, including lung, breast, colorectal, and head and neck cancers. In a phenomenon termed oncogene addiction, N.S.C.L.C. tumours require mutant, E.G.F.R.S for proliferation and survival. Erlotinib, gefitinib, and afatinib are, for E.G.F.R. Tyrosine Kinase Inhibitors approved by the U.S. Food and Drug Administration (F.D.A.) as first-line therapy for N.S.C.L.C. with, E.G.F.R. mutations. The initial clinically significant decrease in tumour burden reflects the attribute of oncogene addiction to EGF and forms the basis of the dramatic response to the EGFR TKI therapy. Although after a median of 12 months, patients who initially demonstrated a good response to, for E.G.F.R. inhibitors usually experience slow tumour progression due to resistance. This resistance is caused due to a second-site mutation T790M at exon 20 in approximately one-half of total cases. Skin toxicity, colitis and rare but potentially fatal is pneumonitis are the main toxicities commonly associated with, E.G.F.R. inhibitors. On C.T. scan this pneumonitis is seen as multifocal ground-glass opacities with or without interlobular septal thickening or diffuse ground-glass densities with consolidation and traction bronchiectasis and are most often misdiagnosed as tumour spread in patients with N.S.C.L.C. The anti-EGFR–directed therapies have been effectively used in other solid malignancies, such as colorectal, head and neck, and breast cancer for instance anti-EGFR antibodies such as cetuximab and panitumumab are presently approved to treat colorectal cancer and head and neck squamous cell cancer. Infusion reaction and rash are a few of the toxicities seen with the anti-EGFR monoclonal antibodies. The acneiform rash seen with cetuximab correlates with a greater response rate in carcinoma treatment. Trastuzumab is a humanized antibody against the extracellular domain of EGFR/HER2 which is commonly seen in breast, gastric and gastroesophageal junction cancers and it causes down-regulation of the intracellular PI3K and M.A.P.K. signalling pathways thereby activating the immune response. Other HER2-targeting agents utilized in carcinoma are pertuzumab, trastuzumab-emtansine, and lapatinib. Pertuzumab inhibits dimerization at HER2/HER3. Pertuzumab shows improved blockade of the HER signalling pathway when combined in treatment with trastuzumab. Cardiotoxicity associated with trastuzumab manifests on imaging as cardiomegaly, pleural effusions and interlobular septal thickening. Imatinib is a small-molecule T.K.I. inhibiting KIT. Toxicities of imatinib manifest on imaging as fluid retention which is seen as ascites, pleural effusions, pulmonary oedema and anasarca. Oncoproteins and Cell Growth Oncogenes have multiple roles, but virtually all encode constitutively active oncoproteins that participate in signalling pathways that regulate the proliferation of cells. Thus protooncogenes, the normal regulated versions of oncogenes, may encode growth factors, growth factor receptors, signal transducers, transcription factors or cell cycle components. Insensitivity to Growth-Inhibiting Signals Tumour suppressor proteins function to control a series of checkpoints responsible for preventing uncontrolled growth. Many tumour suppressors, such as R.B. and p53, constitute a regulatory network that recognizes genotoxic stress from any source and responds by halting proliferation. The protein products of tumour suppressor genes thereby function as transcription factors, cell cycle inhibitors, signal transduction molecules, cell surface receptors and cellular response regulators to D.N.A. damage. Mechanism of Action of Major Tumour Suppressor Genes
2.15: Oncoimaging: Basic science to clinical application
Introduction
Growth Factor
Sources
Functions
Epidermal growth factor (EGF)
Activated macrophages, salivary glands, keratinocytes, many other cells
Mitogenic for many cell types: stimulates epithelial cell migration: stimulates formation of granulation tissue
Transforming growth factor-a (TGF-α)
Activated macrophages, keratinocytes, many other cells
Stimulates proliferation of hepatocytes and many other epithelial cells
Hepatocyte growth factor (HGF) (scatter factor)
Fibroblasts, stromal cells in the liver, endothelial cells
Enhances proliferation of hepatocytes and other epithelial cells: increases cell motility
Vascular endothelial growth factor (VEGF)
Mesenchymal cells
Stimulates proliferation of endothelial cells: increases vascular permeability
Platelet-derived growth factor (PDGF)
Platelets, macrophages, endothelial cells, smooth muscle cells, keratinocytes
Chemotactic for neutrophils, macrophages, fibroblasts, and smooth muscle cells: activates and stimulates proliferation of fibroblasts, endothelial cells, and other cells: stimulates ECM protein synthesis
Fibroblast growth factors (FGFs) including acidic (FGF-I) and basic (FGF-2)
Macrophages, mast cells, endothelial cells, many other cell types
Chemotactic and mitogenic for fibroblasts; stimulates angiogenesis and ECM protein synthesis
Transforming growth factor-β (TGF-fJ)
Platelets, T lymphocytes, macrophages, endothelial cells, epithelial cells, smooth muscle cells, fibroblasts
Chemotactic for leukocytes and fibroblasts: stimulates ECM protein synthesis: suppresses acute inflammation
Keratinocyte growth factor (KGF) (i.e., FGF-7)
Fibroblasts
Stimulates keratinocyte migration, proliferation, and differentiation
The genome
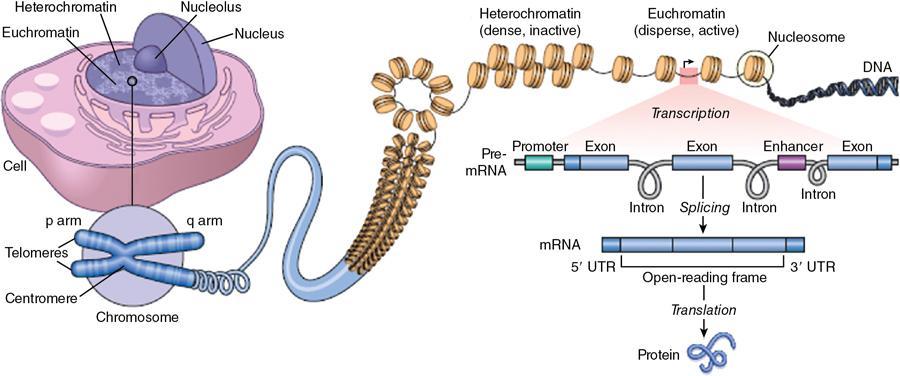
Cell structure and organelles (Fig. 2.15.1.2)
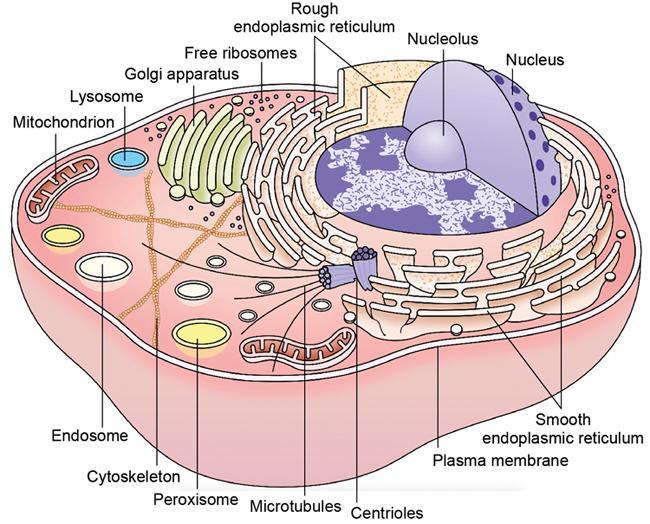
Cell membrane
Cytoskeleton
Cell–cell interactions
Cytoplasm
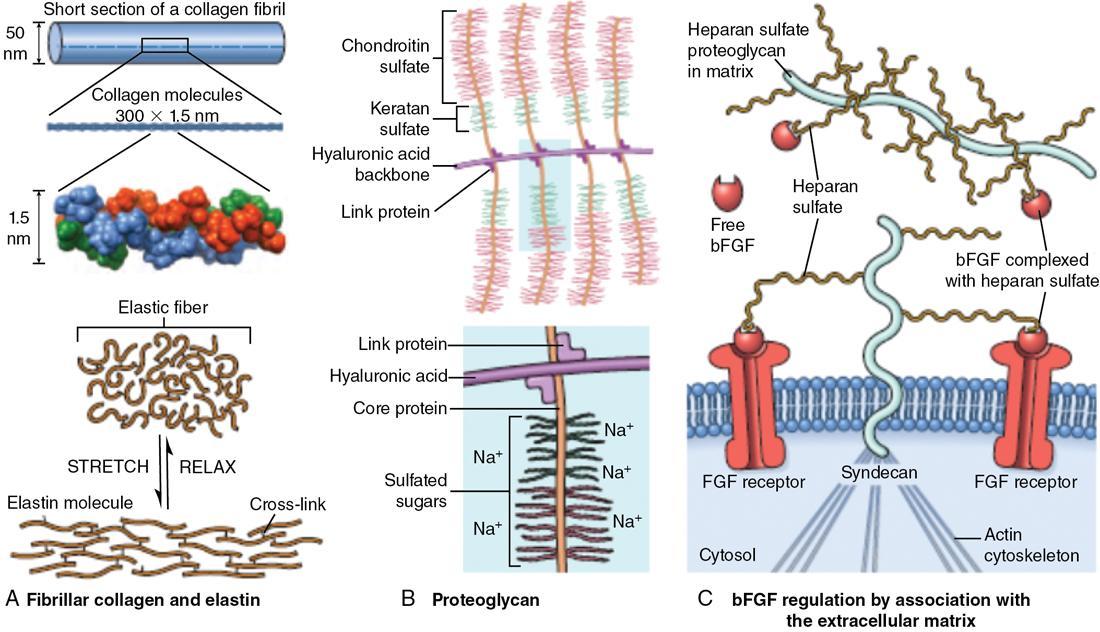
Extracellular matrix
Interstitial matrix and basement membrane
Maintaining cell population
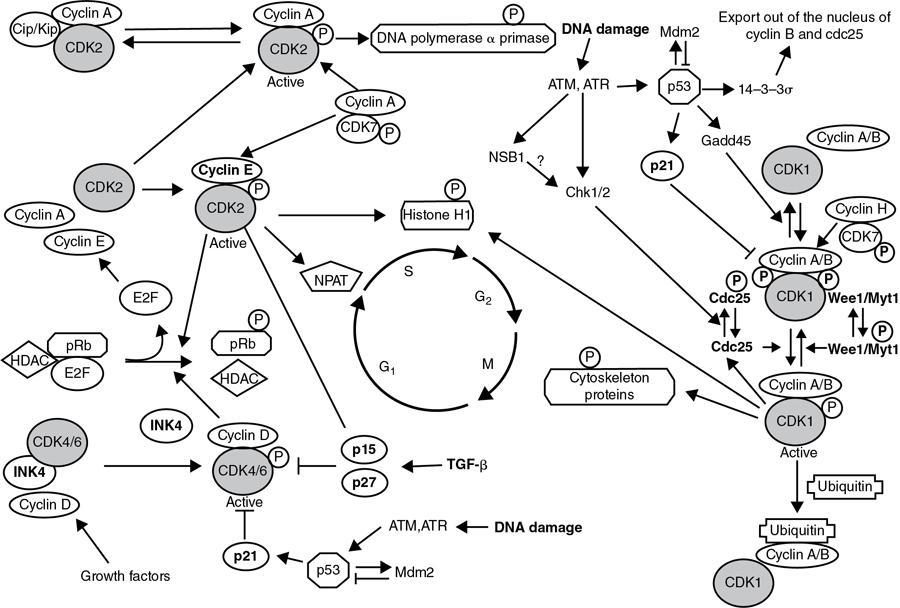
Proliferation and the cell cycle
Stem cells
Regenerative medicine
Introduction to cancer
Nomenclature of cancer
Classification of cancer: Benign vs malignant vs mixed
Tissue of Origin
Benign
Malignant
COMPOSED OF ONE PARENCHYMAL CELL TYPE
TUMORS OF MESENCHYMAL ORIGIN
VESSELS AND SURFACE COVERINGS
TUMORS OF EPITHELIAL ORIGIN
MORE THAN ONE NEOPLASTIC CELL TYPE—MIXED TUMORS, USUALLY DERIVED FROM ONE GERM CELL LAYER
MORE THAN ONE NEOPLASTIC CELL TYPE DERIVED FROM MORE THAN ONE GERM CELL LAYER—TERATOGENOUS
Cancers can be further characterized in terms of
Molecular basis of cancer: Role of genetic and epigenetic alterations
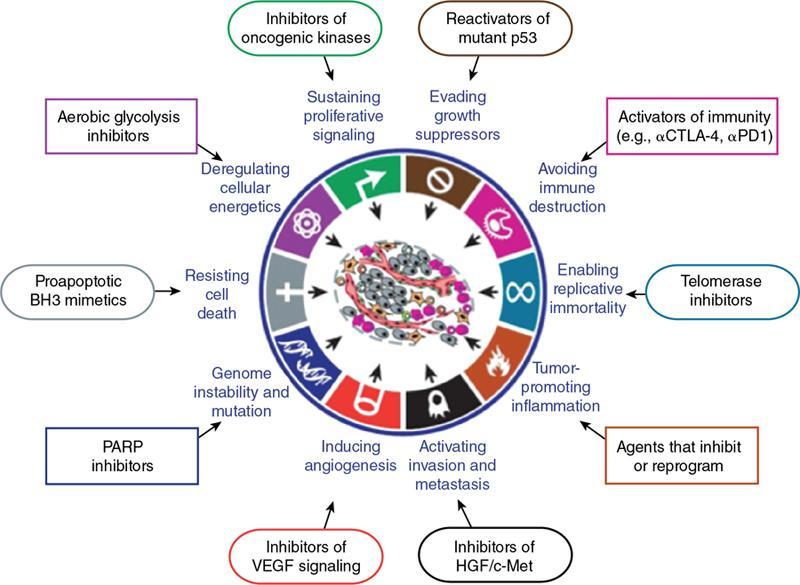
Cellular and molecular hallmarks of cancer (Fig. 2.15.1.4)
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


