CHAPTER 55 Osteogenesis Imperfecta
Epidemiology
Osteogenesis imperfecta (OI) is a genetic disorder that results in deficient fibroblastic and osteoblastic activity. This connective tissue disorder is caused by an error in type I collagen with resultant clinical manifestations that affect not only bone formation but also skin, teeth, and other connective tissue. In greater than 90% of cases, OI results from mutation in one of two genes: the COL1A1 gene on chromosome 17 and the COL1A2 gene on chromosome 7, which encode the chains of type 1 collagen.
Clinical Features
There are four major types of OI with a variable degree of clinical manifestations. Type I is the most common and mildest form of the four and type II is the most severe form. The typical manifestations are a tendency of bone to fracture with minimal trauma, blue sclerae, and deafness. Other clinical features include faulty dentin production, joint dislocations, easy bruisability, and hyperflexibility. In type II, multiple in utero fractures of the skull, vertebrae, and chest wall may result in death in utero or shortly after birth.
Pathology
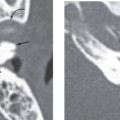
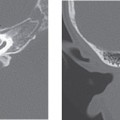
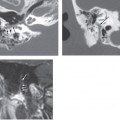
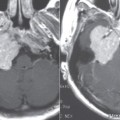
Histopathologic examination of specimens removed from OI patients can provide clues to the type of hearing impairment that can be expected in these patients. In type II patients, there is markedly delayed and deficient ossification in the three layers of the otic capsule with microfractures, deformities, and dehiscence involving the otic capsule and middle ear ossicles. Conductive hearing loss in these patients is mainly due to fractures that commonly involve the crus of the stapes or the handle of the malleus. These fractures can lead to discontinuity of the ossicular chain or to fixation, by ankylosis, of the head of the malleus to the medial attic wall. Adult-onset hearing loss with OI, therefore, can be a conductive type and is not always progressive.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


