Fig. 1
The AR-LBD used as an example to represent location of alternative binding sites of NRs. Standard view of AR-LBD followed by − 90° turn on x-axis and + 90° turn on the y-axis showing both the BF-3 and the AF-2 pockets respectively. These two alternative sites are then showed in detail complexed to small molecule TRIAC as well as a LxxLL/FxxFF motif peptide. AF-2 and BF-3 site are shown in green and pink respectively first as a cartoon representation and then as a surface representation. The hormone DHT is depicted in brown as spheres. The TRIAC molecule (depicted in purple) is shown in stick form while the LxxLL/FxxFF peptide (light green and orange, respectively) is shown as a cartoon representation with the side chains in stick form. Hydrogen bond interactions of the molecules with AR are shown in black dotted lines
The LBD of NRs has also been shown to feature another hydrophobic exposed groove, also conserved across NR subclasses and susceptible of pharmaceutical attack, called binding function 3 (BF-3) . The BF-3 pocket was unexpectedly discovered by X-ray crystallography in the AR and then confirmed through transcriptional assays and site-directed mutagenesis (Estébanez-Perpiñá et al. 2007a). This concave shaped pocket is located adjacent to the AF-2 groove and comparable in size and depth; furthermore some residues are common to both pockets. BF-3 is composed of H1, H3, the loop between H3 and H4 and H9 and unlike the AF-2 pocket, it does not possess any opposite charge cluster delimiting residues. Mutations in the BF-3 site have been shown in AR to greatly boost its activity suggesting that AR-BF-3 site may be a co-repressor site, although this is still to be verified for AR. Although BF-3 may be regulated by protein binders, its most striking characteristic is that of its interconnection with the AF-2 surface conformation and its role in modulating AF-2 capabilities to engage in contacts with coactivator peptides/proteins. Therefore we could say that BF-3 has been shown to modulate coactivator recruitment via allosteric communication with AF-2 (Estébanez-Perpiñá et al. 2007b; Grosdidier et al. 2012).
3 AF-2 Pocket—Possible Drug Target?
Over the years the disruption of NR-cofactor interactions through targeting the AF-2 function has become a more substantiate target area for pharmaceutical intervention. X-ray crystallographic studies revealed that despite the high sequence homology of NRs AF-2 function, NRs can present different electrostatic characteristics and surface shapes that may be exploited to achieve selectivity. Experiments proved that although most coactivators bind thanks to the LxxLL motif, selectivity of peptide sequences between NRs is dependent significantly on the residues flanking the core motif (Vaz et al. 2009; Teichert et al. 2009; Geistlinger and Guy 2003). Different classes of LxxLL interacting motifs from distinct NRs were elucidated thanks to the studies using large focused combinatorial phage libraries. Peroxisome proliferator-activated receptor (PPAR)-LBD, for example, showed preference for HPLxxLL as a small consensus motif, which aided in the discovery of a peptide (NBM131: sequence ATTPPTLHPLLTQFLRTD) (Mettu et al. 2007). This small peptide was found to have a high affinity for both PPARα (NR1C1) and PPARγ (NR1C2) and to compete with coactivators such as PPARγ-coactivator 1α (PGC1α) and repress PPAR mediated activity, opening a new window into PPAR inhibition. Additional studies with other NRs such as the mineralocorticoid receptor (MR/NR3C2) showed that the small motif MPLxxLL has a high affinity for this receptor and that not surprisingly 50 % of all peptides that have been shown to bind to MR to date possess this sequence in their protein sequence. ER-LBD peptide screens with LxxLL core flanked with 7 random amino acids revealed 3 distinct classes of LxxLL interacting proteins (class I: “X5SRLxxLLX7” class II: “X4 H/XPLLxxLLX7 and class III: “X5 S/T L/ILxxLLX7”) that were able to mimic the interaction between ER and endogenous coactivators and block ER transcriptional activity. From these experiments the peptide #293 (SSIKDFPNLISLLSR) was shown to mimic the interaction between ER and physiological coactivators and block ER transcriptional activity exerting a much higher affinity towards the ERβ isoform rather than ERα. (Patent no. WO1999054728A2) (Chang et al. 1999). Recent experiments effectively screened the ER surface for novel peptide binders and found a series of proline peptide sequences with the highly evolved PXLxxLLXXP sequence. This sequence consensus reminiscent of the class II peptides described above, was then biochemically and structurally analyzed. The results obtained proved the use of these flanking prolines residues by the peptide for helix binding optimization to the surface charge clamp by determining the precise helix length. Furthermore, the proline residues seem to increase the hydrophobicity of the charge clamp residues, which strengthens the electrostatic interactions and favors more stabilizing hydrogen bond interactions (Fuchs et al. 2013). A new approach towards NR peptide based inhibition was described by Phillips et al , whereby ER LxxLL based peptides with improved α-helix stability were synthesized by adding a hydrocarbon link, known as stapled peptides (Phillips et al. 2011).
The AR exhibits a peculiarity that is not shared with other NRs. An important feature of the AR LBD is that its AF-2 pocket interacts with its N-terminal domain (NTD), an inter-domain interaction that has key impact in AR physiological actions. AR-NTD contains two NR-like boxes as the ones found in NR coactivators. Surprisingly these motifs contain an FxxLF sequence (residues 23–27 in the AR-NTD, where F stands for phenylalanine) and WxxLF (residues 433–437 in the AR-NTD, W, tryptophan) sequence and they mediate directly the interaction with the AF-2 groove (He et al. 2000 2002; Langley et al. 1995; Wilson 2011; Brodie and McEwan 2005). Both motifs (F/WxxLF and LxxLL) interact in a similar fashion utilizing the charge clamp amino acid K720 for the stabilization and positioning of the α-helix (Vaz et al. 2009). However, a unique feature of AR is its higher affinity for FxxLF motif containing coregulators. This is most likely due to additional H-bonding interactions of this motif with E897 and the malleability of some of the long side chain residues in this binding site that can accommodate bigger residues such as phenylalanine and allow for extra H-bonding interactions (e.g. I737). This is additionally confirmed by the 1 Å shift difference observed between the two classes of regulatory peptides and could be used to our advantage when designing antiandrogen molecules.
Nevertheless, even though all this information obtained by phage display or physiologically-derived peptides have aided in our understanding of the principles of selectivity of these NRs towards their coregulators and ergo their recognition principles, small peptides have limitation in their application in clinical research. It is incredibly challenging to achieve efficient intracellular delivery in vivo of these molecules mostly due to their poor stability and permeability as well as their short plasma half-life.
3.1 Small Molecule Based Inhibitors for AF-2: Can They Be Used in the Clinic?
The earliest example of small molecules known to alter the actions of NRs and that bind outside the LBD were described for ERα. These where pyrimidines that block the interaction of purified estradiol (E2)-activated ERα with labeled SRC1 box 2 peptide in fluorescence polarization assays (Rodriguez et al. 2004) (Table 1). Posterior optimization of these inhibitors led to a pyramidine based inhibitor with an inhibition constant (Ki) value of 2–3 μM using time resolved fluorescence resonance energy transfer (FRET). From then on, many examples of different inhibitors have shown to have effects in vitro and in vivo against ER (Becerril and Hamilton 2007; Zhou et al. 2007). Examples are amphiphatic benzene inhibitors that proved to inhibit mediated transcription of ERα in the endometrial adenocarcinoma HEC-1 cells (Gunther et al. 2009a), or the guanylhydrazone ERI-05 discovered by high throughput screening and that inhibits endogenous expression of the ERα regulated gene pS2 in the breast cancer MCF-7 cell line at a 20 μM concentration (LaFrate et al. 2008; LaFrate et al. 2009) (Table 1). The first crystal structure of ER co-crystallized with one of these coactivator binding site inhibitors was that of 4-hydroxy-tamoxifen (4-OHT) (Fig. 2) (Kojetin et al. 2008; Wang et al. 2006a). Tamoxifen, which has been used for more than 30 years, is one of the most effective treatments for ER-positive breast cancer. This inhibitor, which has been shown to bind to the LBP, had previously hinted that its total binding capacity to ERα was 2 times that of estradiol, the crystal structure ERβ-4-OHT surprised the scientific community by demonstrating that 4-OHT binds both to the AF-2 groove and the expected LBP (Kojetin et al. 2008, Jensen and Khan 2004). The binding of this 4-OHT to the surface appears to be mostly hydrophobic with numerous van der Waals interactions helping to accommodate this inhibitor perfectly in the hydrophobic patches of the coactivator-binding groove (Kojetin et al. 2008; Wang et al. 2006a; Wang 2006b). The binding of 4-OHT to the ER AF-2 pocket may account for the mixed agonist/antagonist activity of type I antiestrogens; nevertheless this discovery opens potential therapeutic avenues for improved antiestrogens.
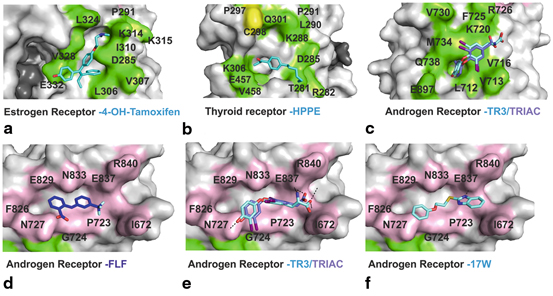
Fig. 2
Surface representations of AF-2 and BF-3 binding sites: residues involved in AF-2 are shown in green while residues involved in BF-3 are depicted in pink. Inhibitors bound to these sites are shown in stick form; carbons are depicted in blue while other element follows the CPK coloring system. a AF-2 binding site of ER co-crystallized with 4-OHT. b AF-2 binding site of TR co-crystallized with HPPE. c AF-2 binding site of AR co-crystallized with inhibitors T3 and TRIAC; AR BF-3 binding site co crystallized with. d FLF e T3 and TRIAC and f compound 32 (2-((2-Phenoxyethyl)thio)-1 H-benzimidazole)
As for ER, the function of most NRs has been modulated through the modulator binding to LBP. This was the case for TR where alterations in function was achieved mostly through analogues of the thyroid hormone (T3), for instance in the treatment of hypothyrodism. The first description of a non-LBP binding inhibitor for TR was that of a β-amino ketone. Arnold et al . carried out a high through put screen (HTPS) utilizing SRC2 box 2 peptide as a fluorescent probe for fluorescence polarization (FP) assays with the TRβ isoform. This HTPS led to a group of α/β unsaturated ketones that were shown to irreversibly bind to an exposed cysteine on the surface of the AF-2 groove of TR, with the best 3-(dibutylamino)-1-(4-hexylphenyl)-propan-1-one (DHPP) having an IC50 of 2 µM (Table 1). Several lines of evidence including TRβ mutants and mass spectroscopy (MS) analysis have led to direct evidence of a covalent adduct formation of these inhibitors to Cys298 in TRβ. This reaction has been shown to occur in two steps, initial beta elimination of the molecule DHPPA to form 1-(4-hexylphenyl)-prop-2-en-1-one (HPPE) followed by a covalent bond formation between HPEE and TRβ. This is also consistent with the fact that a deamination reaction of DHPPA would not be feasible at a physiological pH without the aid of another active molecule. For DHPPA this mechanism is catalyzed directly on the protein surface. X-ray crystallographic studies of DHPPA with TRβ showed the deaminated active principle in the AF-2 groove supporting the specific mechanism of alkylation and deamination in situ catalyzed by Cys298 (Fig. 2b). These compounds were extensively studied and optimized using a structure-activity-relationship (SAR) approach. Though potency measured through FP did not seem to improve other important qualities such as solubility, permeability, cytotoxicity and selectivity towards TR over AR, ERα and PPAR were greatly improved (Arnold et al. 2007; Estébanez-Perpiñá et al. 2007c d; Arnold et al. 2006). Other HTPS have led to the discovery of methylsulfonylnitrobenzoates (MSNB) inhibitors (Hwang et al. 2011). MS and mutational studies demonstrated that these inhibitors also bind to TR irreversibly by targeting the Cys298 residue in the AF-2 pocket. Selectivity towards TRβ over AR and PPARγ as well as good solubility and cytotoxicity profiles made these inhibitors highly interesting. The most potent MSNB inhibitor was MI151 with a potency of 1.8 µM in the FP-SRC2 interaction assay with TRβ showed less off target activity than DPPAH (Hwang et al. 2011). These MSNB inhibitors were further optimized by the removal of the ester linker between the methylsulfonylnitro and the rest of the molecule to a cyclic bioisostere, such a thiazole. These inhibitors were therefore termed sulfonylnitrophenylthiazoles (SNPTs) and showed improved in vivo properties as well as the classical irreversible activity through Cys298 binding. Two inhibitors showed low µM potency, that of 2{4,1,5} and 2{3,1,2} showed by FP-SRC-2 binding assays to have an IC50 of 0.3 and 2.4 µM respectively however the less active of these two presented additionally good transcriptional inhibitory activity (5 µM in the T3-mediated luciferase assay) (Hwang et al. 2012) (Table 1).
Inhibition of NRs through their AF-2 binding pocket has only been minimally attempted in other NRs. Two AF-2 small molecule inhibitors have been described for the vitamin D receptor (VDR/NR1I1) and although there is no in vivo data of their inhibition, they have proven to bind to this NR in vitro by a TR-FRET assay. The first non-LBP binding inhibitors for the VDR were the benzodiazepines derivatives (Mita et al. 2010) (Table 1). These inhibitors proved to affect the interaction between VDR and LxxLL coactivator peptides, whereby the most potent of the group showed an IC50 of 20 µM in a VDR TR-FRET based assay (Mita et al. 2010). Monitoring of the effect of benzodiazepines in VDR mediated transcription, in the presence of increased concentration of its natural ligand (1,25 dihydrozyvitamin D3), showed no shift in the dose response curve, confirming the non-LBP binding nature (Mita et al. 2010). It was then shown that this inhibitor acts as an α-helix peptido-mimetic inhibitor, whereby the three alkyl side chains presented in this inhibitor mimic the three leucines in the LxxLL motif. The tetrahydrobenxodiazepinamine therefore acts as a rigid structure that mimics the α-helical arrangement and the methylbutene and the two isopentane groups mimic the leucines in the “NR box” motif of coactivators (Mita 2010). In a separate study, small molecule 3-indolyl-methanamides were identified as alternative site binders for the VDR LBD alternative binding was that of (Nandhikonda et al. 2012) (Table 1). These molecules block the interaction of VDR with SRC-2 in FP-based assays as well as inhibit VDR mediated transcriptions at a cellular level and presented with good selectivity towards VDR over AR, ERβ, TRα, TRβ and PPARγ (Nandhikonda et al. 2012). Surprisingly this indol-methanamine proved to bind to VDR irreversibly involving a reaction mechanism, whereby an azafulvenium salt is produced as an intermediate compound forming a positive like species, which can act with any nucleophile in the surface of VDR (Nandhikonda et al. 2012). However this promising inhibitor which has an IC50 in the low micromolar range proved to have high cytotoxicity that is yet to be determined if it is due to the irreversible mechanism observed (Nandhikonda et al. 2012). More in vivo data is required in order to see if this inhibitor is a viable option for clinical inhibition of VDR.
Another example of non-LBP bound inhibitors was shown for the NR pregnane X receptor (PXR/NR1I2). PXR, as well as the constitutive androstate receptor (CAR/NR1I3), are two orphan NRs, meaning that no endogenous ligand has been identified as of yet (Willson and Kliewer 2002). However, it has been shown that they can function as xenobiotic sensors as they interact with many foreign chemicals (Biswas et al. 2009). Antagonists for PXR are of great interest due to their impact on the therapeutic efficiency of drugs and their ability to prevent harmful drug-drug interactions. Nevertheless, this is a highly complicated task as their LBP is highly flexible and can accommodate many ligands as has been repeatedly shown in the many structures of PXR co-crystallized with different ligands of different sizes (Xue et al. 2007; Watkins et al. 2001; Chrencik et al. 2005; Cheng and Redinbo 2011). The first non-LBP ligand described for PXR is ketoconazole, a known antifungal drug that is also used as an antiandrogen for metastatic PCa (Peer et al. 2014; Huang et al. 2007) (Table 1). This molecule antagonizes PXR in the presence of the antibiotic rifampicin, a known agonist of PXR and was also shown to disrupt the interaction of activated PXR with SRC-1 in a non-competitive fashion (Huang et al. 2007). Furthermore, the non-LBP binding nature of ketoconazole to PXR was confirmed through scintillation proximity assays, which excluded a potential LBP competitive mechanism. This was further confirmed by site-directed mutagenesis. Other inhibitors have been shown to bind to AF-2 such as the phytochemical coumestrol, found to bind to the surface of PXR in functional gene reporter assays and to antagonize PXR ligand dependent SRC-1 recruitment to the same extent regardless of the concentration of rifampicin (Want et al. 2008). FP-competition experiments with fluorescently labeled SRC-1 LxxLL ligands confirmed its binding to AF-2 (Huang et al. 2007;Wang et al. 2007; Ekins et al. 2007). This inhibitor was found to be highly selective for PXR except for some cross-reactivity with ER. The drug leflunomide is used for rheumathoid arthritis and is another inhibitor which was found to show inhibition of the PXR/SRC-1interaction and proved by site-directed mutagenesis, to directly bind to AF-2 (Ekins et al. 2008). However, more structural studies are needed to view the binding positioning of these inhibitors and additional in vivo data of these inhibitors has not been obtained to date (Ekins et al. 2007 2008).
ER, TR, PXR and VDR are all NRs whose AF-2 function has been shown to be a viable druggable non-LBP target. Nonetheless following ER, AR is the second NR whereby its non-LBP binding pockets have been extensively exploited for rational drug design. The first non-LBP binding inhibitors for AR were based on the pyrimidines inhibitors, initially designed for ER (Gunther et al. 2009b). The authors rationally designed tri-substituted peptidomimetic pyrimidines with bulkier groups than those initially utilized for ER emulating the AR preference for bulkier side chains (i.e FxxLF) in the NR boxes of co-regulatory proteins (Gunther et al. 2009b). This classical peptidomimetic approach achieved pyrimidines that were AR selective over ER and that had IC50 values ranging 1.5–6.6 μM as determined by AR inhibition in a transcriptional activated cellular assay (Gunther et al. 2009b).
Axerio-Cilies et al . carried out a structure-based drug design approach using all the structural information available on AR and its binding to different coactivators (Axerio-Cilies et al. 2011). This study led to a set of molecules with a rigid linker and two aromatic features that mimicked an FxxLF NR box: the diarylhydrazide inhibitors (Axerio-Cilies et al. 2011; Caboni et al. 2012). With high selectivity for AR over GR (NR3C1), ERα/β, but still showing some inhibition of PR, these inhibitors were shown to inhibit AR coactivator binding around 13–26 μM IC50 in a TR-FRET assay. Furthermore, they induce prostate specific antigen (PSA) expression in the absence of androgens and to inhibit DHT and cyproterone acetate stimulated PSA (Axerio-Cilies et al. 2011; Caboni et al. 2012) (Table 1). A further example of the utility of peptidomimetics for AR targeting was published recently by Ravindranathan et al. (2013) . This group evaluated the X-ray structure of peptide coactivator sequence LxxLL/FxxLF bound to AR and designed two compounds mimicking these coactivator motifs. The main structural scaffold was that of a bis-benzamide group where two benzyl groups of compound D1 mimicked FxxLF motif and in the analogous compound D2, two isobutyl groups were used to mimick the LxxLL motif (Ravindranathan et al. 2013). Surprisingly, D1 did not show any inhibitory effect in AR-coactivator binding, however, D2 was shown to block the interaction of many coregulator proteins in co-immunoprecipitation assays using the LNCaP cell line (Ravindranathan et al. 2013). D2 also proved to block AR dependent transcriptional activity and prevent AR nuclear translocation as well as inhibiting AR-mediated PCa cell proliferation. Furthermore, it was reported to inhibit AR-dependent growth of xenograft tumors in vivo and AR expression in human tumors cultured ex vivo (Ravindranathan et al. 2013). These findings provide evidence that targeting the AF-2 binding site of AR using peptidomimetics may be a viable therapeutic approach for patients with advanced PCa.
Other functional FP screens with the aim of identifying potential PCa treatment molecules binding to the AF-2 function identified the off-patent nonsteroidal antiinflamatory drugs (NSAIDs) and thyroid hormones as potential antagonists of AR (Estébanez-Perpiñá et al. 2007a; Buzón et al. 2012). This screen led to the identification of: (i) two kinase inhibitors, 1-tert-butyl-3-(2,5-dimethyl-benzyl-)-1 H-pyrazolo[3,4,-D]pyrimidin-4-ylamine (K10) and 3-((1-tert-butyl-4-amino-1 H-pyrazolo[3,4D]pyrimidin-3-yl)methyl)phenol (RB-1); (ii) five small molecules, flufenamic acid (FLF), tolfenamic acid (TOL), meclofenamic acid (MEL), 2-methylindole and indole-3-carboxylic acid; and (iii) two thyroid hormones, triiodothyronine (T3) and 3,5,3’-triiodothyroacetic acid (TRIAC) (Table 1). The complex structure of the two kinase inhibitors with AR proved their binding preference to the AF-2 binding site. These inhibitors form a plethora of hydrophobic interactions and are additionally stabilized by H-bonding to the residue M734. RB-1 was shown to form an additional H-bond with the residue K720, a key residue that forms part of the “charge clamp” residues that normally stabilize the LxxLL motifs in their binding to the AF-2 site. Some small molecules such as 2-methylindole also showed by crystallographic data to be located in the AF-2 occupying the same subpocket of the AF-2 groove as the pyrimidine moiety of RB-1 and K10. The two thyroid hormones T3 and TRIAC also proved to bind to this coregulator binding groove, however these two inhibitors seem to not form any hydrogen bonds and be stabilized uniquely by week hydrophobic interactions (Fig. 2c). These two thyroid hormone compounds and some other small molecules in this screen proved to additionally bind to the exposed BF-3 pocket (Fig. 2e).
4 A Neighboring Pocket Emerging as a Possible Drug Target Site–The BF-3
Additionally to those AF-2 binding molecules identified in the FP-screen by Estébanez-Perpiñá et al . and contrary to what was expected, the X-ray crystallographic data did not reveal the TRIAC or T3 uniquely in the AF-2 groove but showed an additional strong uniformly well defined electron density in another novel surface pocket named BF-3 (Estébanez-Perpiñá et al. 2007a). Furthermore, the NSAID called flufenamic acid (FLF) was shown to be uniquely bound to the BF-3 pocket of AR (Fig. 2d). Moreover, the TRIAC and T3 inhibitor proved to interact preferentially at the BF-3 binding site versus in Cursive the AF-2. In the BF-3 these inhibitors form many hydrophobic interactions and H-bonds with the residues N727 and E837, strongly securing the inhibitors in place (Fig. 2e). In the case of T3 an additional hydrogen bond is observed between the carboxyl group of T3 and the protein backbone and side chain of residue I672. On the other hand, the small molecule FLF showed uniquely hydrophobic interactions with the BF-3 surface (Fig. 2d) (Estébanez-Perpiñá et al. 2007a). Since the discovery of the BF-3 pocket many other AR inhibitors have been identified to bind to this non-LBP binding site. De Leon et al . elucidated a lead compound, termed MJC013, a dichloropheyl-cyclohexanecarboxamide molecule, which inhibits AR function by preventing hormone dependent dissociation of the Hsp90-FKBP52-AR complex and that uniquely binds to the BF-3 binding site in AR, as shown by functional studies (De Leon et al. 2011).
Furthermore, recently a large virtual screen, performed by Lack et al . using the Zinc database and AR LBD crystal structure, lead to the identification of several different scaffolds of inhibitors that proved to inhibit AR both in eGFP screening assays and in AR transcriptional luciferase assays using LNCaP and HeLa cells (Irwin et al. 2012; Irwin and Shoichet 2005; Lack et al. 2011). The highly selective compounds 1–4 (ZINC03445992/ZINC03877300/ZINC02058890 and ZINC00012342 respectively) were analyzed by means of X-ray crystallography , which proved that all these four inhibitors bound to BF-3 except compound 1 which was shown to be bound to BF-3 and AF-2 (Lack et al. 2011) (Table 1). Compounds 1 and 3 were shown to be stabilized mainly by hydrophobic interactions; meanwhile compounds 2 and 4 formed additionally hydrogen bonding interactions with residue N727, a key residue of both the AF-2 and BF-3 pockets. This work proved the possibility of successfully screening for inhibitors incapable of repressing AR transcriptional activity and binding to the BF-3 pocket, going from 10 million commercially available compounds to 8 possible candidates. However further in vivo data of these inhibitors is required. From this extensive report the same group carried out a further optimization of a set of compounds elucidated, with a core thiol-benzimidazole structure (Lack et al. 2011; Munuganti et al. 2013). Based on the previous crystal data obtained, the authors decided to remove the 2-(4-methylphenoxy)ethanamine group, a moiety of the compound which did not seem to form any essential interactions for stabilization in the BF-3 pocket. This led to a functional core for rational drug design, the 2-[(2-Phenoxyethyl)thio]− 1 H-benzimidazole (compound 32), which inhibited AR transcriptional activity 3-fold stronger (IC50 4.2 μM) (Munuganti et al. 2013). The crystal structure showed a well defined electron density and positioned the inhibitor in the BF-3 binding site where it makes van-der-Waals interactions as well as a strong hydrogen bond with E837 (Munuganti et al. 2013). Various SAR studies were performed with compound 32, in order to obtain improved derivatives; replacement of the oxygen atom in SC2H4O did not improve the potency of the inhibitor, however addition of small hydrophobic substituents in the benzene ring was able to enhance anti-AR potency leading to two compounds in the low μM range (compound 47 and 49 containing methyl at meta and diortho positions respectively). Furthermore, these compounds did not only prove to be specific for AR, compared to ERα, but to additionally exhibit effective growth inhibition in both LNCaP and ENZ-resistant cell lines (Munuganti et al. 2013). All these inhibitors confirm that this BF-3 orientated compounds show a different anti-AR action to clinically used anti-androgens and with little cell toxicity, however in vivo studies are still required in order to fully understand the physiological effect of inhibiting BF-3 pocket.
All of these BF-3 targeting inhibitors here presented have been designed to alter AR function; however the in-depth analysis of these structures as well as other NRs X-ray structures proved that this BF-3 pocket is conserved amongst different NRs (Estébanez-Perpiñá et al. 2007a; Buzón et al. 2012). Opening a new targeting site for non-LBP inhibitors, nevertheless little is known on the structural/functional relationship of NRs binding to coregulators, mutational studies in the BF-3 site hint at the possibility of the BF-3 surface allosterically communicating with the AF-2 groove influencing coactivator recruitment as a consequence (Estébanez-Perpiñá et al. 2007a).
5 Communication and Allosteric Mechanisms Between AF-2 and BF-3
As previously mentioned AF-2 can undergo subtle induced fit rearrangements upon coactivator binding and several residue side chains move to create a larger hydrophobic pocket that will accommodate the bulky hydrophobic side chains, like in the case of the AR that can accommodate W/FxxLF motifs present in the AR-NTD and AR specific coactivator such as ARA70. Several of the compounds mentioned above, including TRIAC and members of the fenamic acid series of anti-inflammatory compounds, although having been shown to preferentially bind to BF-3, still inhibit AR/LxxLL, which occurs in the AF-2 groove (Grosdidier et al. 2012). In the case of AR, crystal structures with and without TRIAC suggests that compounds binding to BF-3 trigger allosteric alterations that propagate to AF-2 and inhibit coregulator binding (Estébanez-Perpiñá et al. 2005b; Grosdidier et al. 2012). All these results suggest allosteric communication between the two pockets and have been complemented by many mutational studies (Estébanez-Perpiñá et al. 2005b; Grosdidier et al. 2012; Buzón et al. 2012). The biggest characterization of this AF-2/BF-3 allosteric communication has been undertaken for the AR. Mutations of residues in the interface of these two pockets (Fig. 1), such as N727K, have been shown to make the AR behave as a ‘super AR’ variant. Surprisingly, mutants such as N833R, which is 6.8 Å away from NR727K, and R840A, which is 22.0 Å away from NR727K, show the same results. Additionally, other mutations such as R840E totally abolish AR LBD activity in vitro while albeit maintaining its 3D structure or only moderately enhance (F826L/N727K) or inhibit (F826R/R726L) activity (Estébanez-Perpiñá et al. 2007a; Grosdidier et al. 2012). These mutations in BF-3, distant from AF-2 surface, have additionally been shown to have an effect on AR-LBD activation by GRIP1 and N/C interdomain interaction, however with a completely different pattern, which further supports the effect of these mutations in the structural rearrangement of AR (Askew et al. 2011). This connecting network is of great importance due to the effect that mutations in BF-3 have in coregulator binding that has been observed to lead different diseases, such as that of PCa (E670, Ile672, L830) and androgen insensitivity syndromes (AIS) (L830, P723, E724, R840) in AR (Estébanez-Perpiñá et al. 2007d). However, further studies must be undertaken in order to fully understand this intra-domain mapping network that NRs have repeatedly demonstrated to have, proving its plasticity and environmental adaptability.
6 LBD Dimerization Interface—Interesting Approach?
Most NRs have been shown to exert their transcriptional function as a homo- and/or hetero-dimers. This ability to dimerize is essential for their function and usually occurs initially in the LBD through the H9 of one monomer and H10/H11 of the other. However, different NRs differ slightly from each other’s dimerisation mechanism, allowing for a specific/individual targeting of these proteins using this alternative area of the LBD. For example in the NR ERR, which has three different isoforms ERRα/β/γ (NR3B1/NR3B2/NR3B3), ERRγ homodimerization and enhances transactivation while heterodimerization with ERRα inhibits transcriptional activities of both ERRα and ERRγ (Gearhart et al. 2003; Gerhard 2004).
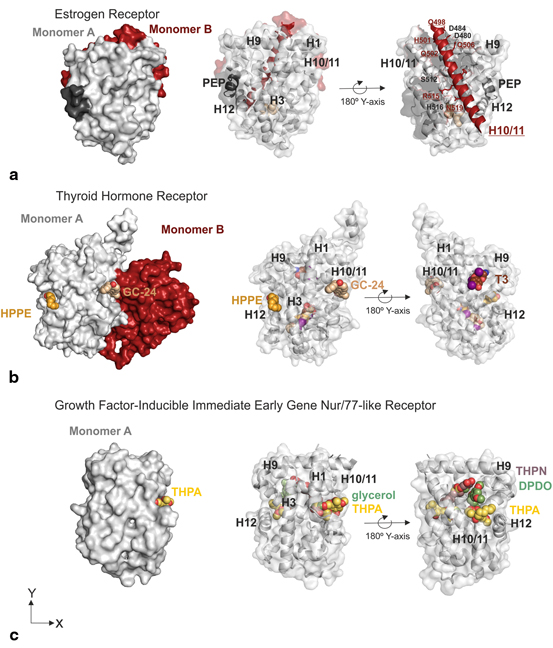
Although at embryonic stages, dimerisation targeting has been attempted in order to block ER function. Chakraborty et al . presented an extensive study of ERα dimerisation characterization in silico using the X-ray structure of ERα-LBD bound to the ligand DES (PDB: 3ERD) (Chakraborty et al. 2013). H-bonding contacts between the two dimers occur mainly from the N-terminal portion of H10/H11 (these helices are not separated in ER) and the H9 of the other monomer. Three main regions of contact have been described; region I (DKITD-monomer A with QQQHQRLAQ-monomer B), region II (QQQHQRLAQ-monomer A with DKITD-monomer B) and region III (LSHIRMMSNK-monomer A with LSHIRMMSNK-monomer B). These regions are unaltered by the presence or absence of DES and show a high polar environment with distinct patches of positive/negatively charged regions. YP537 (sequence CNVVPLYPDLLLE) was the first peptide to inhibit ER without binding to AF-2 groove (Arnold and Notides 1995). It was then suggested that this inhibitor binds as a “dimerisation inhibitor”. This peptide was shown to bind in the human ER dimerisation site and precipitate independently to hormone bound or unbound ER (apo state as well as when ER was incubated E2, 4-OHT and ICI182,780). However it must be mentioned that protein misfolding and aggregation are not likely to be the best option in a therapeutic context due to functional in vivo consequences not being straightforward and that NRs are generally bound to chaperones in their inactive form in order to prevent misfolding. Other in silico experiments have identified an extended and a mutant version of the binding sequence motif region II a foremost mentioned, the LQQQHQQLAQ sequence which has been proposed to inhibit dimerisation in liganded and un-liganded ERα (Chakraborty et al. 2012).
Evidence of the dimerisation interface as a possible drug target was also observed for TRβ. The crystallographic data of the inhibitor, GC-24, with the receptor LBD contained an electron density for GC-24 in the LBP, but also an additional one for a second GC-24 molecule, placed in the potential dimer interface of TR (Fig. 3) (Borngraeber et al. 2003). The additional molecule is clearly seen bound in a surface pocket formed by H1, H9, H10 and H11 in the opposite site of the AF2 (Fig. 3b). This fact is consistent with the fact that the crystallization conditions utilized by the group were different to those previously described by others, as these did not yield any crystals (Borngraeber et al. 2003). A recent paper also reported a new X-ray crystallography structure of TRα-LBD that depicted a second binding site for T3 and T4, TR ligands, located between H9, H10 and H11 forming H-bonding with residues Q342, E339 and R375. These two pockets have also been observed in the NR growth-inducible immediate early gene nur/77-like receptor (Nur77) (NR4A1). A recent screen of a Nur77 targeting compounds led to the discovery of 1-(3,4,5-trihydroxyphenyl)nonan-1-one (THNP) and its derivative 1-(3,5-dimethoxyphenyl)decan-1-one (DPDO) (Souza et al. 2014; Wang 2014). THNP has recently been shown to have an integral role in melanoma autophagic cell death via Nur77 (NR4A1) inhibition. The complex structure of THNP depicted this inhibitors bound between H9, H10 and H11, an area that the authors term Site C (Wang et al. 2014). An overlay of the THNP-Nur77 structure with that of TR-T3 complex proved both these ligands to be bound in the same area. Surprisingly THPN and not DPDO was shown to facilitate the interaction with Nur77 through the binding to this site C with leads to the formation of tumors in the liver and lung in a Nur77 dependent manner (Wang et al. 2014). Comparison of both crystal structures of THPN and DPDO proved that although both these ligands bind between H10-H11 and H9, DPDO interacts at a location close to but not at site C which may explain the inability of DPDO to form the LBD-interacting surface for Nix that results in the lack of DPDO-associated Nur77 targeting to mitochondria and melanoma cell death (Wang et al. 2014).
 Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
 Estrogen Receptor Modulators (SERMs) and Selective Androgen Receptor Modulators (SARMs)
Estrogen Receptor Modulators (SERMs) and Selective Androgen Receptor Modulators (SARMs)
 Analyses of Ordered and Disordered Regions in Ecdysteroid Receptor
Analyses of Ordered and Disordered Regions in Ecdysteroid Receptor
 and Regulation of Nuclear Receptor Corepressor Complexes
and Regulation of Nuclear Receptor Corepressor Complexes
 Receptor Structure and Function
Receptor Structure and Function
 Analysis of Heterodimeric Nuclear Receptors
Analysis of Heterodimeric Nuclear Receptors




Related posts:
Multi-Functional Androgen Receptor Coregulator and Proto-Oncogene Melanoma Antigen-A11 (MAGE-A11)
Estrogen Receptor Modulators (SERMs) and Selective Androgen Receptor Modulators (SARMs)
Analyses of Ordered and Disordered Regions in Ecdysteroid Receptor
and Regulation of Nuclear Receptor Corepressor Complexes
Receptor Structure and Function
Analysis of Heterodimeric Nuclear Receptors
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
