Paget Disease
Pathophysiology
Paget disease, a relatively common bone disorder, is a chronic, progressive disturbance in bone metabolism that primarily affects older persons. It is slightly more common in men than in women (3:2), with an average age of onset between 45 and 55 years, although the disease has been known to occur in young adults. The prevalence of Paget disease varies considerably in different parts of the world, reaching its greatest incidence in Great Britain, Australia, and New Zealand.
The precise nature of Paget disease and its etiology are still debatable. Sir James Paget named the disease osteitis deformans in the belief that the basic process was infectious in origin. Other etiologies have also been proposed, such as neoplastic, vascular, endocrinologic, immunologic, traumatic, and hereditary. The hereditary etiology was supported by identification of mutations in the gene encoding sequestosome 1 (SQSTM1/p62) in patients with familial and sporadic Paget disease. Recent ultrastructural studies and the discovery of giant multinucleated osteoclasts containing microfilaments in the affected cytoplasm, as well as intranuclear inclusion bodies, suggest a viral etiology. Some investigators have obtained immunocytologic evidence identifying the particles as analogous to those from the measles group virus material. Other immunologic studies have demonstrated viral antigens in affected cells identical to those from the respiratory syncytial virus. The most recent research indicates a paramyxovirus as an etiologic factor.
Whatever the fundamental cause of Paget disease, its basic pathologic process has to do with the balance between bone resorption and appositional new bone formation. There is disordered and extremely active bone remodeling, secondary to both osteoclastic bone resorption and osteoblastic bone formation in a characteristic mosaic pattern, which is the histologic hallmark of this condition. Biochemically, the increase in osteoblastic activity is reflected in elevated levels of serum alkaline phosphatase, which can rise to extremely high values. Similarly, the increase in osteoclastic bone resorption is reflected in high urinary levels of hydroxyproline, which is formed as a result of collagen breakdown.
The skeletal abnormalities seen in Paget disease are frequently asymptomatic and may be an incidental finding on radiographic examination or at autopsy. When the changes are symptomatic, clinical manifestations are often related to complications of the disease, such as deformity of the long bones, warmth in the involved extremity, periosteal tenderness and bone pain, fractures, secondary osteoarthritis, neural compression, and sarcomatous degeneration. The distribution of a lesion varies from monostotic involvement to widespread disease. The following bones, in order of decreasing frequency, are most often affected: the pelvis, femur, skull, tibia, vertebrae, clavicle, humerus, and ribs (Fig. 29.1). The fibula is involved only in exceptional cases.
Radiologic Evaluation
The radiographic features of Paget disease correspond to the pathologic processes in the bone and depend on the stage of the disorder. In the early phase, the osteolytic or hot phase, active bone resorption is evident as a radiolucent wedge or an elongated area with sharp borders that destroys both the cortex and cancellous bone as it advances along the shaft. The terms frequently used to describe this phenomenon are “advancing wedge,” “candle flame,” and “blade of grass” (Fig. 29.2). In flat bones such as the calvarium or the iliac bone, an area of active bone destruction known as osteoporosis circumscripta appears as a purely osteolytic lesion (Fig. 29.3). In the skull, most commonly affected sites are the frontal and occipital bones; both inner and outer calvarial tables are involved, but the former is usually more extensively affected.
In the intermediate or mixed phase, bone destruction is accompanied by new bone formation, with the latter process tending to predominate. Bone remodeling appears radiographically as thickening of the cortex and coarse trabeculation of cancellous bone (Fig. 29.4). In the pelvis, cortical thickening and sclerosis of the iliopectineal and ischiopubic lines are present. Pubic rami and ischia may enlarge. In the spine, the thin cortex of the vertebral body, which disappears in the hot phase, is later replaced by broad, coarsely trabeculated bone, forming what appears to be a “picture frame” around the body (Fig. 29.5). In the skull, focal patchy densities with a “cotton-ball” appearance are characteristic (Fig. 29.6).
In the cool or sclerotic phase, a diffuse increase of bone density occurs together with enlargement and widening of the bone and marked cortical thickening, with blurring of the demarcation between cortex and spongiosa (Fig. 29.7). Bowing of long bones may become a striking feature (Fig. 29.8). Similar changes are observed in the skull, where obliteration of the diploic space is also a typical feature (Fig. 29.9).
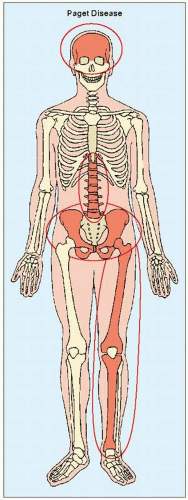 FIGURE 29.1 Major target sites of Paget disease. |
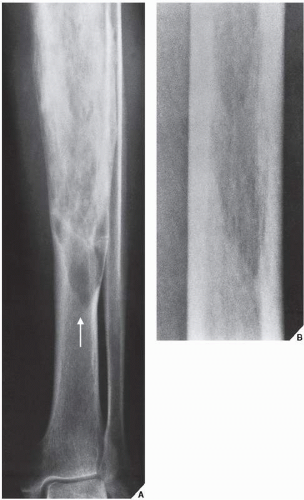 FIGURE 29.2 Osteolytic phase of Paget disease. (A) Anteroposterior radiograph of the lower leg of a 68-year-old woman shows an advancing wedge of osteolytic destruction in the midportion of the tibia (arrow). (B) Magnification study of the midfemur in another patient shows the purely osteolytic phase of Paget disease. In both examples, the lesion resembles a blade of grass or a candle flame. (A from Sissons HA, Greenspan A, 1986, with permission.) |
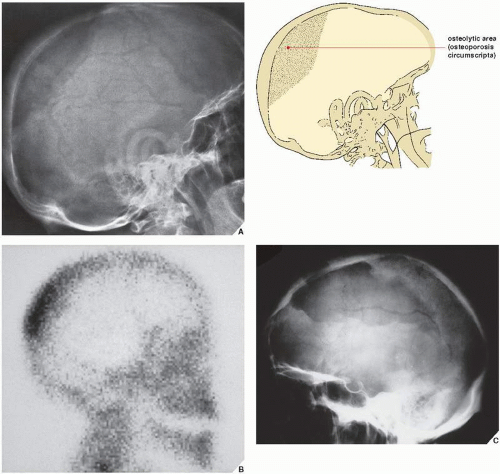 FIGURE 29.3 Osteolytic phase of Paget disease. (A) Lateral radiograph of the skull of a 60-year-old man shows an osteolytic lesion in the parieto-occipital area. This sharply demarcated defect, known as osteoporosis circumscripta, represents a hot phase of the disease. (B) Radionuclide bone scan shows a characteristic localized increased uptake of the radiopharmaceutical tracer resulting in the appearance of a “yarmulke” sign. (C) Lateral radiograph of the skull of a 65-year-old woman reveals osteoporosis circumscripta in the fronto-parietal area. |
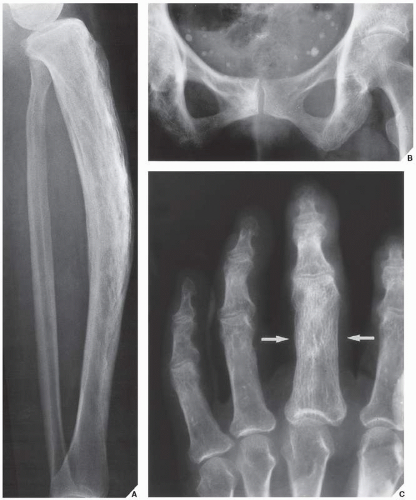 FIGURE 29.4 Intermediate phase of Paget disease. (A) In the intermediate phase, seen here affecting the tibia in a 62-year-old woman, thickening of the cortex and a coarse trabecular pattern in the medullary portion of the bone are characteristic features. Note the anterior bowing. (B) In another patient, an 81-year-old woman, intermediate phase is seen in the pubic and ischial bones. (C) Mixed phase affecting the proximal phalanx of the middle finger (arrows) is seen in a 67-year-old woman with monostotic Paget disease. |
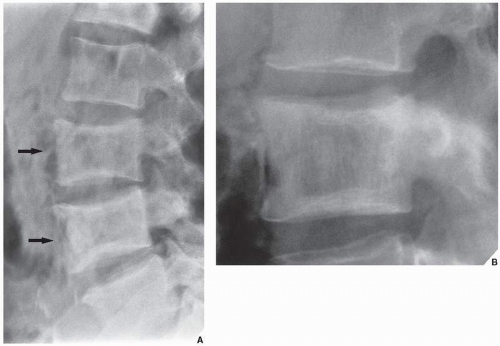 FIGURE 29.5 Intermediate phase of Paget disease. (A) Involvement of the lumbar spine in the mixed phase can be recognized by the “picture frame” appearance of the vertebral bodies (arrows) created by dense sclerotic bone on the periphery and greater radiolucency in the center. Note the partial replacement of vertebral end plates by coarsely trabeculated bone. (B) In another patient, the “picture frame” appearance of the vertebral body of L2 marks the intermediate phase of Paget disease. (A from Sissons HA, Greenspan A, 1986, with permission.) |
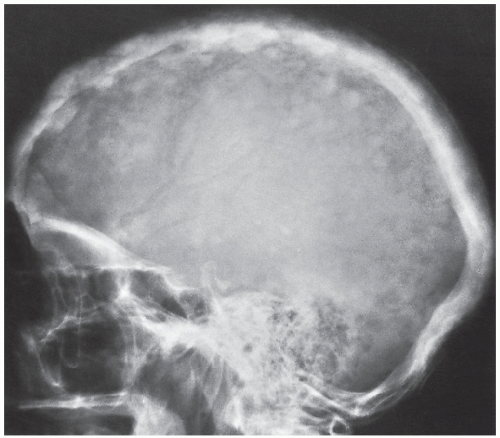 FIGURE 29.6 Intermediate phase of Paget disease. Focal patchy densities in the skull, having a “cotton ball” appearance, are typical of the intermediate phase of Paget disease as seen in this radiograph of a 68-year-old woman. |
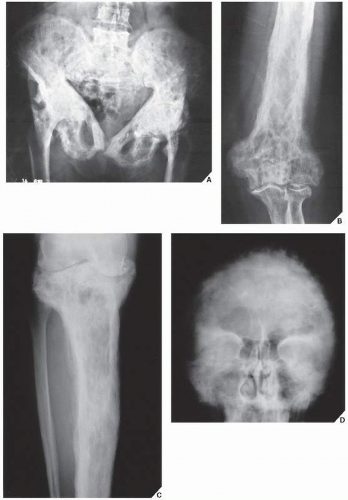 FIGURE 29.7 Cool phase of Paget disease. In the cool phase, there is considerable thickening of the cortex and bone deformity. (A) The pelvic cavity, seen here in an 80-year-old woman, may assume a triangular appearance. (B) Involvement of a long bone, in this case the distal humerus of a 60-year-old woman, exhibits marked cortical thickening, narrowing of the medullary cavity, and a coarse trabecular pattern. (C) Similar changes are present in the tibia in a 72-year-old man. (D) Anteroposterior radiograph of the skull of an 82-year-old woman reveals typical changes of the cool phase of Paget disease. (A and B from Sissons HA, Greenspan A, 1986, with permission.) |
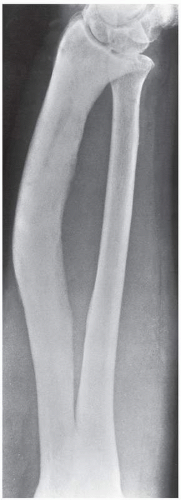 FIGURE 29.8 Cool phase of Paget disease. Anteroposterior radiograph of the forearm of a 57-year-old man with polyostotic Paget disease shows enlargement of the left radius with a marked bowing deformity. Other signs of the cool phase of the disease are seen in the diffuse sclerotic changes and the indistinct demarcation between the cortex and the spongiosa. |
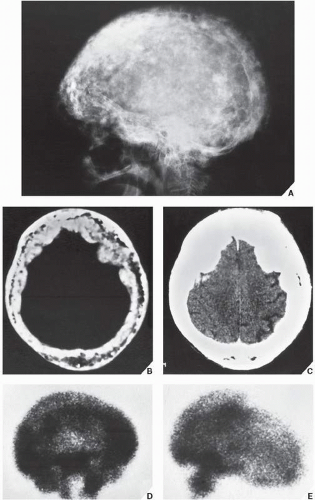 FIGURE 29.9 Cool phase of Paget disease. (A) Lateral radiograph of the skull of an 80-year-old woman demonstrates numerous coalescent densities associated with thickening and sclerosis of the cranial vault and base of the skull. CT sections clearly demonstrate predominant involvement of the inner table with marked diminution of the diploic space (B) and thickening of the cranial vault (C). (D,E) Scintigraphy demonstrates markedly increased uptake of radiopharmaceutical. |
It is important to remember that, since in the long bones Paget disease starts at one articular end and advances to the other, all three phases of the disorder may coexist in the same bone (Fig. 29.10A). Likewise, different phases may coexist in the flat bones or in the spine (Fig. 29.10B).
Computed tomography (CT) may demonstrate characteristic features of Paget disease (Fig. 29.11), although it is rarely required. Magnetic resonance imaging (MRI) is occasionally employed to demonstrate cortical and intramedullary involvement better, and to exclude (or confirm) extension of the process into the soft tissues. In general, the pagetic bone exhibits heterogeneous signal intensity. On T1-weighted sequences, intermediate-to-low signal intensity is usually noted. On T2 weighting, the signal may be high, intermediate, or low, depending on the stage of the disease and degree of fibrosis and sclerosis (Figs. 29.12 and 29.13).
Differential Diagnosis
Several conditions may mimic Paget disease, while the disease itself may be mistaken for other pathologic processes; for example, involvement of a single bone can be mistaken for monostotic fibrous dysplasia, and a uniform increase in osseous density may mimic lymphoma or metastatic cancer. The rugger-jersey appearance of the spine in secondary hyperparathyroidism may resemble Paget vertebra (see Fig. 28.8). Vertebral hemangioma also looks very much like Paget vertebra on a radiograph, except that the vertebral body is not enlarged and the vertebral end plates are well outlined (see Fig. 20.46). However, the condition that bears the most striking resembalance to Paget disease is familial idiopathic hyperphosphatasia, also called “juvenile Paget disease” (see Figs. 30.1 and 30.2). In this condition, unlike Paget disease, the articular ends of the bone may not be affected.
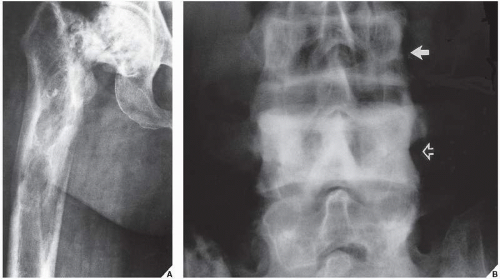 FIGURE 29.10 Coexistence of different phases of Paget disease. (A) Anteroposterior radiograph of the proximal half of the femur of a 77-year-old woman demonstrates all three phases of the disorder. The cool phase is seen in the femoral head, the intermediate phase in the proximal shaft, and the hot phase, represented by an osteolytic wedge of resorption, in the medial cortex more distally. (B) In another patient, a 54-year-old man, intermediate phase is seen in the vertebra L3 (arrow), whereas the L4 reveals a cool phase (open arrow). |
Complications
Pathologic Fractures
Of the numerous complications observed in patients with Paget disease, the most common are pathologic fractures in the long bones. They may resemble partial or incomplete stress fractures, appearing radiographically as multiple short horizontal radiolucent lines on the convex aspect of the cortex (Fig. 29.14). True complete fractures are referred to as “banana-type” because of the horizontal direction of the fracture line as it traverses the affected bone (Fig. 29.15), and they have also been compared with crushed rotten wood or chalk. Fractures are more likely to occur during the osteolytic or hot phase, and they are frequently the main presenting manifestation of Paget disease.
Related posts:
 Miscellaneous Metabolic and Endocrine Disorders
Miscellaneous Metabolic and Endocrine Disorders
 Benign Tumors and Tumor-like Lesions III: Fibrous, Fibroosseus, and Fibrohistiocytic Lesions
Benign Tumors and Tumor-like Lesions III: Fibrous, Fibroosseus, and Fibrohistiocytic Lesions
 Upper Limb III: Distal Forearm, Wrist, and Hand
Upper Limb III: Distal Forearm, Wrist, and Hand
 Benign Tumors and Tumor-Like Lesions IV: Miscellaneous Lesions
Benign Tumors and Tumor-Like Lesions IV: Miscellaneous Lesions
 Upper Limb III: Distal Forearm, Wrist, and Hand
Upper Limb III: Distal Forearm, Wrist, and Hand
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


