Typical angina
1. Substernal chest discomfort with a characteristic quality and duration
2. Worse with exertion or emotional stress
3. Relieved by rest or nitroglycerin
Atypical angina: meets two of the characteristic above
Noncardiac chest pain: meets ≤1 of the characteristic above
Table 3.2
Canadian cardiovascular society grading of angina pectoris
Grade 1 | Ordinary physical activity does no cause angina, such as walking and climbing stairs. Angina with strenuous or rapid or prolonged exertion at work or recreation |
Grade 2 | Slight limitation of ordinary activity. Walking or climbing stairs rapidly, walking uphill, walking or stair climbing after meals, or in cold, in wind or under emotional stress, or only during the few hours after awakening. Walking more than two blocks on the level and climbing more than one flight of ordinary stairs at a normal pace and in normal conditions |
Grade 3 | Marked limitation of ordinary physical activity. Walking one or two blocks on the level and climbing one flight of stairs in normal conditions and at normal pace |
Grade 4 | Inability to carry out any physical activity without discomfort; angina may be present at rest |
Stable angina (also called typical angina) is the most common form of angina pectoris and is caused by an imbalance of coronary perfusion relative to myocardial oxygen demand. Stable angina is usually relieved by factors that decrease myocardial oxygen demand (such as rest) or increase coronary perfusion (such as vasodilator) [2].
Unstable angina (USA) is type of angina that has pattern of increasingly frequent pain, longer duration, and change in quality and is a warning sign that an acute MI may be imminent. USA of most often caused by disruption of an atherosclerotic plaque, partial thrombus formation, embolization, and/or coronary vasospasm. The characteristic features of USA include: (1) Occurs are rest or with minimal exertions, (2) New onset within 1 month, and (3) Occurs with a crescendo pattern (more severe, prolonged, or increased frequency than previously) [2–4].
Prinzmetal variant angina is an uncommon form myocardial ischemia caused by coronary vasospasm. It is important to note that Prinzmetal angina may occur in patients with or without significant coronary atherosclerosis. Multiple etiologies have been identified as the cause of coronary artery spasm and include smoking, cocaine use, and stress although many cases occur spontaneously without any identifiable cause. The clinical presentation of Prinzmetal angina is characterized by chest discomfort that occurs at rest (rather than on exertion) thus patient may exhibit a circadian pattern of symptoms with episodes occurring at night or during the early morning hours [2, 3, 5].
Definition: Myocardial Infarction
Myocardial infarction is defined as a clinical event caused by ischemia in which there is evidence of myocardial injury or necrosis. Evidence of ischemia is met when there is a rise and fall of cardiac biomarkers with supportive evidence including patient symptoms, ECG findings, and/or imaging findings (such as wall motion abnormality on an echocardiogram). Elevated troponins due to myocardial injury may be due to multiple causes included primary myocardial ischemia, supply/demand ischemia, myocardial injury not related to ischemia, or multifactorial (Table 3.3). In 2012 the Third Universal Definition of MI was release and is outlined in Table 3.4.
Table 3.3
Causes of elevated cardiac troponins
Myocardial injury related to primary coronary artery injury |
Plaque rupture |
Coronary artery thrombus formation |
Coronary vasculitis |
Myocardial injury related to supply/demand mismatch |
Coronary spasm |
Severe hypertension or hypotension |
Tachy/brady – arrhythmia |
Severe aortic stenosis |
Severe anemia |
Respiratory failure |
Coronary emboli |
Myocardial injury not related to ischemia |
Myocarditis |
Cardiac contusion |
Defibrillator shock |
Surgery |
Cardiotoxic drugs |
Multifactorial |
Takotsubo cardiomyopathy |
Sepsis |
Heart failure |
Pulmonary emboli |
Infiltrative diseases |
Table 3.4
Universal classification of myocardial infarction
Type 1: Spontaneous myocardial infarction |
Spontaneous myocardial infarction related to atherosclerotic plaque rupture, ulceration, erosion, or dissection with resulting intraluminal thrombus in one or more of the coronary arteries leading to decreased myocardial blood flow or distal platelet emboli with myocyte necrosis. The patient may have underlying severe CAD but on occasion non-obstructive or no CAD |
Type 2: Myocardial infarction secondary to an ischemic imbalance |
In instances of myocardial injury with necrosis where a condition other than CAD contributes to an imbalance between myocardial oxygen supply and/or demand, e.g. coronary endothelial dysfunction, coronary artery spasm, coronary embolism, tachy-/brady-arrhythmias, anemia, respiratory failure, hypotension, and hypertension with or without LVH |
Type 3: Myocardial infarction resulting in death when biomarker values are unavailable |
Cardiac death with symptoms suggestive of myocardial ischemia and presumed new ischemic ECG changes or new LBBB, but death occurring before blood samples could be obtained, before cardiac biomarker could rise, or in rare cases cardiac biomarkers were not collected |
Type 4a: Myocardial infarction related to percutaneous coronary intervention (PCI) |
Myocardial infarction associated with PCI is defined by elevation of troponins >5 times the upper limit of normal in patients with normal baseline values or a rise of troponins by 20 % if the baseline values are elevated and are stable or falling |
In addition, one for the following must be present: |
Symptoms suggestive of myocardial ischemia |
New ischemic ECG changes or new LBBB |
Angiographic loss of patency of a major coronary artery or a side branch or persistent slow or no-flow or embolization |
Imaging demonstration of new loss of viable myocardium or new regional wall motion abnormality |
Type 4b: Myocardial infarction related to stent thrombosis |
Myocardial infarction associated with stent thrombosis is detected by coronary angiography or autopsy in the setting of myocardial ischemia and with a rise and/or fall of cardiac biomarkers values with at least one value above the upper limit of normal URL |
Type 5: Myocardial infarction related to coronary artery bypass grafting (CABG) |
Myocardial infarction associated with CABG is defined by elevation of cardiac biomarker values 10 times the upper limit of normal in patients with normal baseline troponin values |
In addition, one for the following must be present: |
New pathological Q waves or new LBBB |
Angiographic documented new graft or new native coronary artery occlusion |
Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality |
Commonly the clinical diagnosis and treatment of a myocardial infarction is based on ECG findings as a means of distinguishing between two types of MI, one that is marked by ST elevation (STEMI) and one that is not (NSTEMI). The terms transmural and nontransmural (subendocardial) MI are no longer used because ECG findings in patients with this condition do not closely correlated with pathologic changes in the myocardium. The presence of Q waves or ST elevation is associated with higher early mortality and morbidity; however, the absence of these two findings does not confer better long-term mortality and morbidity [4, 6].
Coronary Artery Anatomy
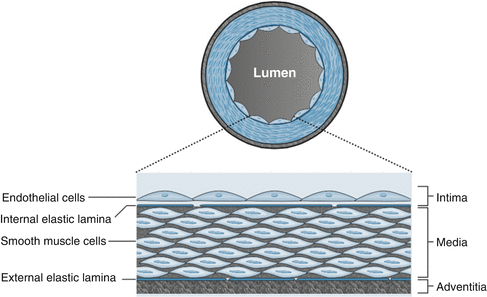
As discussed in Chap. 1, the normal coronary artery wall consists of three layers called the intima, media, and adventitia (See Fig. 3.1). The intima is separated from the media by a thin membrane call the internal elastic membrane. The media is separated from the adventitia by a thin membrane call the external elastic membrane. The intimal layer is closest to the arterial lumen and consists of a single layer of cells called the endothelium. The endothelium is a highly metabolically active barrier of tissue that serves multiple functions with the most notable being maintenance of hemostasis and immune cell recruitment. The media is the thickest layer of tissue and contains mostly smooth muscle cells as well as the extracellular matrix. The media enable the vessel to dilate and constrict in order to control blood flow. The adventitia is the outermost layer and consists mostly of fibro-elastic tissue. The adventitia is more prominent in larger vessels such as the aorta and is primary branches. The function of the adventitia is to stretch and recoil in response to systolic pressures produced by the heart.
Atherosclerosis
The most common cause of heart disease and CAD is caused by atherosclerosis. “The term atherosclerosis is derived from the Greek word “athero”, meaning gruel, or wax, corresponding to the necrotic core area at the base of the atherosclerotic plaque, and “sclerosis” for hardening, or induration, referring to the fibrous cap of the plaque’s luminal edge” [7]. Atherosclerosis is a slowly progressive chronic disease that is due to the build up of lipid within the artery wall resulting in thickening and hardening of the vessel. The development of atherosclerosis is asymptomatic until it reaches an advanced stage or is triggers by an acute cardiovascular event.
Atherosclerotic CAD is the leading cause of morbidly and mortality worldwide [8] and is responsible for about one in every six deaths in the United States [9]. The economic burden of cardiovascular disease (CVD) is rapidly growing compromising the national health care expenditure and cost the US economy over $100 billion each year. It is estimated that by 2030 40 % of the US population will have some form of CVD [10].
Theories of Atherogenesis
Several theories have been proposed over the past two centuries to explain the pathogenesis and development of atherosclerotic plaques. As our knowledge has rapidly increased we have found that the individual proposed theories are not mutually exclusive, and are linked to each other. Leonardo da Vinci (1452–1519AD) was one of the first to document the macroscopic findings of an atherosclerotic artery when he illustrated the artery of a human autopsy specimen and suggested that the thickening of the vessel wall might be due to “excessive nourishment” [11]. In the nineteenth century a physician by the name of Carl von Rokitansky documented the macroscopic findings of multiple plaques in the aorta. He hypothesized that degenerated blood bound proteins deposit into the arterial wall and formed pseudomembranes that had both thrombogenic and calcific characteristics [12]. Several years later Rudolf Virchow (1821–1902) built upon his work by studying microscopic sections of diseased vessels. Virchow concluded that atherosclerotic lesions were located in the intimal layer of blood vessels and that despots within this layer was associated with connective tissue proliferation resulting in vessel wall degeneration [13]. In the early twentieth century several scientist contributed to identifying the content of arterial plaques. The most notable was a German scientist by the name of Adolf Windaus who won a Nobel prize in 1910 after showing that cholesterol was present in atherosclerotic lesions [14]. Throughout the nineteenth century as people began to live longer, interest grew to determine the clinic implications cause atherosclerosis. Several theories were proposed during this time, and as our understanding of the pathophysiology of atherosclerosis increased, we have recognized that process is of increasing complexity with a multitude of variables. Below we will outline several of the most pertinent theories of atherosclerosis.
Insudation Theory
This theory proposes that the primary event that triggers the development atherosclerosis is focal accumulation of lipids in the vessel wall. The lipids from these lesions are believed to be derived from plasma lipoproteins (i.e. low density lipoproteins, LDL), which is consistent with the role of hyperlipidemia being a risk factor for atherosclerosis and acute coronary syndrome. The LDL particle is too large to penetrate the tightly closed junctions between adjacent endothelial cells. However, endothelial cells have receptors for LDL and are able to transport these lipids across an intact endothelium by receptor-mediated uptake. Lipids may also be engulfed by monocytes while in the circulation and then transport into the intima as they transmigrate into the wall between dysfunctional endothelial cells. Lipids that are in the extracellular space within the intima become trapped within the extracellular matrix where they accumulate over time.
Encrustation Theory
This theory initially proposed that material from the blood was deposited on the inner surface of arteries and lead to thickening of the inner lining creating a mural thrombus. It was thought that platelets and fibrin of thrombi initiated atherosclerosis. Although mural thrombosis is not the initial event in atherogenesis, it is likely to promote the later progression of the atherosclerotic lesion and is the major event leading to vascular occlusion, especially in coronary arteries.
Reaction to Injury Theory
The reaction to injury theory is the most widely excepted among scientific and medical scholars. This theory states that the initial event in the pathogenesis of atherosclerosis is injury to the endothelium that compromises the integrity of the endothelial barrier. The nature of the injury is likely to involve numerous factors and is different in different people depending on genetic and environmental conditions. Injured endothelium triggers an inflammatory response with migration of inflammatory cells (predominantly macrophages and lymphocyte). Inflammatory cell migration along with oxidized low-density lipoproteins within the lesion is thought to be the key player during the development of an atherosclerotic plaque [13, 15].
Atherosclerosis: Pathophysiology
Microscopic Description of the Development of an Atherosclerotic Lesion
Endothelial injury and dysfunction is the cornerstone for the initiation of atherosclerosis. The specific mechanism contributing to endothelial dysfunction is not completely understood, although the most important culprits that trigger endothelial dysfunction include hypertension, hyperlipidemia, diabetes, and toxin from cigarette smoking. Regardless of the cause of endothelial dysfunction this lead to increased vascular permeability.
In the presence of a lipoprotein abnormality lipids begin to accumulate on the endothelial cells lining the endothelium. As the lipid particles accumulate they migrate into the intima and coalesce into the proteoglycan of the extracellular matrix (ECM) were they get “trapped” (Fig. 3.2). Lipoproteins that accumulate in the ECM are susceptible to oxidative stress. The oxidized lipoproteins produce free radicals that causes localize cellular injury and dysfunction. This stimulates the release of growth factors, cytokines, and chemokines by endothelial cells, and hinders the vessels vasodilator activity.
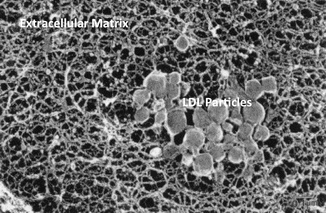
Fig. 3.2
Electron micrograph image of aorta from a experimental animal that received intravenous injection of human low-density lipoprotein (LDL). Round LDL particles seen trapped in the extracellular matric of the intima (From Douglas et al. [3]. with permission)
Accumulation of extracellular lipoprotein particles is one of the first morphologic changes to occur. Lipoprotein particles become trapped in the strands of proteoglycan within the subendothelial region of the intima where they begin to accumulate. Proteoglycan-associated lipoproteins appears particularly are susceptible to oxidative modification. In response to oxidized lipoproteins, cellular injury occurs and triggers an immune respond with the recruitment of monocytes that migrate into the intima of the artery wall (Fig. 3.3). Once monocytes have migrated into the intima they differentiate into macrophages and engulf the harmful oxidized lipoproteins form intracellular lipid filled vacuoles. These macrophages that are filled with vacuoles of lipoproteins are referred to as foam cells. Although, the foam cells serve a theoretically protective function the oxidized lipoproteins within these cells augments cellular activation and cytokine production resulting in additional recruitment of mononuclear cells. Activate macrophages also produce reactive oxygen species that further augments oxidation of lipoproteins within the cell. Over time the macrophages and foams cells die releasing the lipid content into the intercellular space. As a result of this cyclic chronic inflammatory state (with local tissue injury, endothelial dysfunction, and breakdown of the vessel wall endothelial barrier), T lymphocyte recruitment occurs resulting in additional release of cytokines that further stimulates macrophages, endothelial cells, smooth muscle cells, and proliferation of the extracellular matrix. With the accumulation of foam cells and the proliferation of intimal smooth muscle cells and extracellular matrix the earliest grossly visible lesion sign of an atherosclerotic lesion develops called a fatty streak. Over time the lesions further mature into an atheroma and will progressively grow with the development of a lipid core. During the lesion maturation process the smooth muscle cells synthesizing extracellular matrix (notably collagen) in an attempt to stabilize the atherosclerotic plaques. Many times this is unsuccessful resulting in an unstable plaque that is prone to rupture [2].
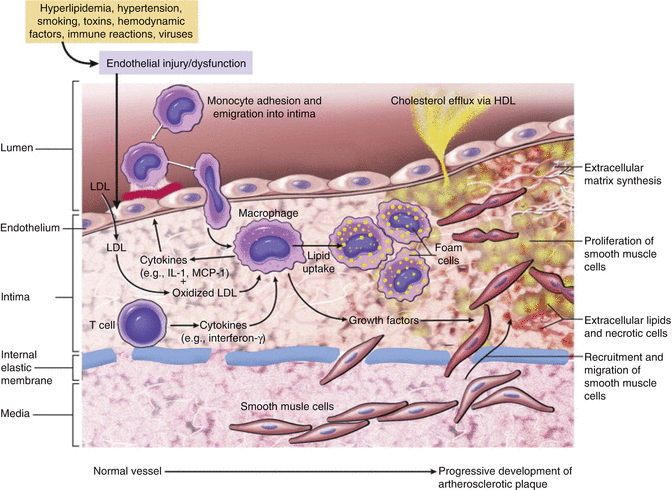
Fig. 3.3




“Hypothetical sequence of cellular interactions in atherosclerosis. Hyperlipidemia and other risk factors are thought to cause endothelial injury, resulting in adhesion of platelets and monocytes and release of growth factors, which leads to smooth muscle cell migration and proliferation. Extracellular lipid is derived from insudation from the vessel lumen, particularly in the presence of hypercholesterolemia, and also from degenerating foam cells. Smooth muscle cells migrate to the intima, proliferate, and produce extracellular matrix” (From Vinay et al. [2] with permission)
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
