Fig. 31.1
Sagittal contrast-enhanced T1-weighted MRI scan showing a large cystic craniopharyngioma
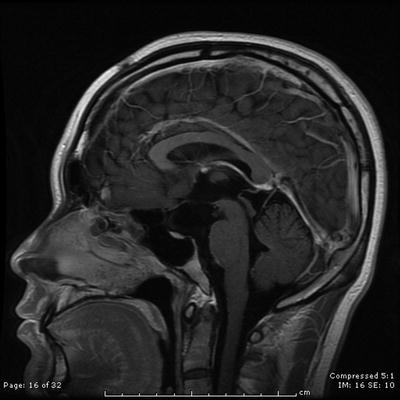
Fig. 31.2
On last follow-up 1 year after Gamma knife radiosurgery for residual tumor, the patient remains stable and the tumor controlled
Neurofibromatosis-1 (NF-1)
Neurofibromatosis-1, or von Recklinghausen NF, is one of the most common autosomal-dominant CNS disorders with an incidence of about 1 per 2,500–3,000 live births [28, 29]. About half of all cases are inherited; the other half arise de novo. The most common genetic abnormality associated with NF results in the loss of a tumor suppressor gene. NF-1 patients are prone to the development of pilocytic astrocytomas and gliomas involving the optic pathway in children [30]. An estimated 15–20 % of children with NF-1 are afflicted with an optic pathway glioma (OPG), with a majority of symptomatic children diagnosed before the age of 6 [3, 31, 32]. Rodriguez et al. [30] reported that in NF-1 patients with pilocytic astrocytomas and indeterminate subtype low-grade astrocytoma, extent of resection was not significantly associated with recurrence, radiographic progression, or tumor location. However, patients who undergo GTR have a better overall survival (OS) rate than those who undergo STR or biopsy (100 % vs. 52 % and 65 % at 10 years, respectively) [30]. Chemotherapy is considered a first-line therapy in children with NF-1 and afflicted with low-grade gliomas. A Children’s Oncology Group phase III trial of 401 children treated with different chemotherapy regimens revealed 5 year OS rates of 98 % for patients with NF-1 vs. 86 % for non-NF-1 children, suggesting a more favorable prognosis in the context of NF-1 [33]. Radiation therapy in patients with NF-1 is limited due to the increased risk of second malignancies [34] and radiation-related cerebral vasculopathies [35]. Sharif et al. [34] reported a high incidence of second nervous system tumors in NF1 OPG patients, with an increased risk in irradiated patients (4 malignant peripheral-nerve sheath tumors in 12 irradiated NF-1 OPG patients vs. 3/48 NF-1 controls).
Neurofibromatosis-2 (NF-2)
Neurofibromatosis-2, or bilateral acoustic NF, is more rare than NF-1 with an incidence of 1 in 25,000 live births [36–38]. Though NF-2 is often associated with adult onset, Evans et al. [39] showed that at least 18 % of NF-2 patients present in childhood and are likely to have a more severely debilitating disease course. Furthermore, 10–18 % of children presenting with a meningioma or schwannoma are likely to have NF-2 with prolonged follow-up [39]. Eighth nerve dysfunction is a much more common symptom in children than in adults. Nunes and MacCollin [40] reported that hearing loss owing to vestibular tumors affected 9/12 children and 11/24 ears by age 18 years. Treatment for NF-2 is primarily surgical; optimal management includes presymptomatic resection for vestibular schwannomas [40]. Vestibular schwannomas in NF-2 patients are more difficult to treat than those with sporadic unilateral VS given the association with the eighth cranial nerve (vestibulocochlear) and as such surgery almost always leads to loss of the cochlear nerve and total deafness [41]. Depending on the location of the tumor, surgical resection via a middle fossa or retrosigmoid approach with partial or complete tumor removal may preserve hearing [42]. SRS is a controversial treatment of vestibular schwannomas in NF-2 and is currently being extensively debated [43]. While radiosurgery may preserve hearing function and arrest tumor growth, the presence of acoustic neuroma is still of threat to the patient [42].
Meningioma
Meningiomas are rare in children, accounting for 0.4–4.6 % of all CNS tumors in the pediatric population [44, 45]. As previously discussed, meningiomas within the pediatric age group are often the first sign of neurofibromatosis 2 [46]. Pediatric meningiomas are most commonly intracranial (90 %), followed by intraspinal (6 %), intraorbital (2 %), and elsewhere (2 %). These tumors are frequently located in the cerebral convexity, followed by intraventricular, falcine, parasagittal, anterior, and middle cranial fossa [47]. Perry et al. [48] noted that meningiomas in the pediatric population have a high frequency of brain invasion and are more phenotypically and genotypically aggressive when compared to adults. Surgical management is difficult in children due to location of tumors, larger size at presentation, relatively less blood volume in children, and risks inherent in long operations such as massive blood transfusions and hypothermia [49]. The goal of treatment is considered gross total resection with resection of the dural origin or attachment recommended to lower the risk of recurrence [45, 50]. In the largest series of 87 pediatric patients by Rushing et al. [50], mortality rate and recurrence rate were reported as 10.6 % and 19.4 %, respectively. There is limited data on the use of SRS in children, reported only in case reports. In one such report, SRS was used to successfully treat a child with recurrent meningioma in association with meningioangiomatosis [51]. Recurrent meningioma has also been successfully treated in three children using Gamma knife radiosurgery [52, 53].
High-Grade Tumors
Treatment for high-grade tumors without brain stem involvement in children is generally approached by surgical resection followed by adjuvant therapy. The main goals of surgery for these malignant tumors include obtaining a histological diagnosis and reducing the burden of the tumor by decreasing mass effect. High-grade tumors that have the treatment option of stereotactic radiosurgery following surgical resection include glioblastomas, ependymomas, medulloblastomas, ependymoblastomas, pineoblastomas, and CNS primitive neuroectodermal tumors (PNETs). GTR is achieved less for eloquent brain areas such as the thalamus and the basal ganglia than noneloquent areas such as the superficial cortical locations not associated with essential functions (63 % vs. 91 %, respectively) [54].
Glioblastoma
Glioblastomas are the most malignant astrocytic neoplasm and characteristically have zones of coagulation necrosis and areas of vascular hyperplasia [55]. These tumors represent 3 % of CNS tumors in children with a 5-year survival of about 20 % [56]. Due to the aggressive nature of glioblastomas, prognosis is poor; the median survival of these patients is only 12 months [55]. Glioblastomas are more commonly found in the superficial cortex than in deep structures [57], and often as a consequence of intratumoral hemorrhage, approximately 5–10 % of these children present as the result of sudden deterioration in their neurological status [58]. In order to significantly increase overall survival of the patient, gross total resection should be the goal. However, this may be more feasible in children than adults. In a study of 37 pediatric patients with glioblastoma not of the brainstem, Yang et al. [57] reported a median OS of 45.1 months in patients with GTR vs. <1 year in patients who had STR or biopsy only. Furthermore, at 5 years, 42 % of patients who had GTR were still alive, whereas none of the patients without GTR survived [57]. Extent of resection but not location of the lesion was reported as an independent predictive factor of OS in a multivariate analysis performed by Song et al. [59]. Though radiation and chemotherapy exist as current treatment options, glioblastomas have an inherent resistance of conventional therapy. Therefore, SRS is thought to be more advantageous due to its improved set-up accuracy in treating recurrent glioblastomas [60]. However, to date there is no prospective study reviewing the efficacy of SRS in pediatric patients with glioblastoma.
Ependymoma
Ependymomas are the third most common intracranial tumors in children, representing up to 10 % of all CNS tumors in the pediatric age range with a peak incidence between birth and 4 years [61]. The greatest determinant of survival in children with ependymoma is extent of resection. Supratentorial ependymomas, constituting about 30 % of ependymomas in children [62], have a better prognosis than infrantentorial ependymomas [63–65]. GTR of supratentorial ependymomas is more likely to be achieved when compared to those in the infratentorial location [66, 67], especially those which do not invade the ventricular system [68, 69]. In these patients with supratentorial ependymomas who undergo complete resection and exhibit no evidence of metastasis, surgery alone may be acceptable treatment option and radiation therapy can be avoided without affected OS, as evidenced by Venkatramani et al. [68].
Gross total resection has been found to dramatically increase OS and PFS. In one study at Children’s Hospital Los Angeles, 28 children with a mean age of 43 months (range 5–51 months) were surgically treated for ependymomas in a 10-year period [61]. Twenty-one (75 %) patients underwent GTR, whereas 7 (25 %) underwent STR or biopsy. In a median follow-up period of 30 months (range 1–95 months), the 28 patients had 3- and 5-year PFS rates of 21.4 % and 10.7 %, respectively, and 3- and 5-OS rates of 39.3 % and 17.9 %, respectively. Patients receiving radiographically confirmed GTR had a much higher PFS and OS (33 and 44 months, respectively), than those receiving STR (6.5 and 15 months, respectively), as shown in Figs. 31.3 and 31.4. Following maximal safe resection, postoperative MRI should be performed to radiologically confirm the extent of resection. If there is residual tumor found with no dissemination, second-look surgery is strongly advised. Even with maximal resection, however, recurrence rates may be as high as 50 %.
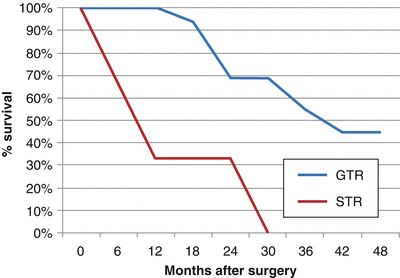
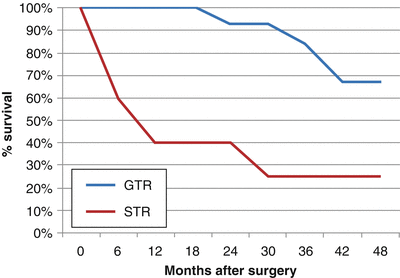
Fig. 31.3
Percent progression-free survival vs. months after surgery for ependymoma patients following GTR and STR
Fig. 31.4
Percent overall survival vs. months after surgery for ependymoma patients following GTR and STR
Adjuvant radiation therapy of 54–55.8 Gy to the tumor bed is a commonly accepted postsurgical treatment for children over the age of 3 years old. One series from UCSF showed that radiation therapy has been found to improve PFS even after adjusting for extent of resection and also interestingly in those who received GTR when compared to those without radiation therapy [70]. The adverse effects of radiation on children under 3 years of age have traditionally limited this type of treatment, though a series by Koshy et al. [71] used data from Surveillance Epidemiology and End Results of 184 children younger than 3 years to show that postoperative radiation therapy significantly improved 3-year overall survival when compared with those who did not receive RT (81 % and 58 %, respectively, p = 0.005). Grundy et al. [72] reported in his series of 89 children younger than 3 years that primary postoperative chemotherapy led to a 5-year OS of 63.4 %, without radiation therapy in 42 % of those treated for nonmetastatic disease. Currently under clinical evaluation is a randomized phase III trial studying how well maintenance chemotherapy works in comparison with observation following induction chemotherapy and radiation therapy in treating newly diagnosed pediatric ependymoma.
Patients with recurrent ependymomas may also want to consider stereotactic radiosurgery. In evaluating the utility of SRS in treating childhood ependymomas, two factors must be considered: efficacy in disease control and toxicity [73]. In 2008, Merchant et al. [74] described their experience with reirradiation for childhood ependymomas at St. Jude’s Hospital. Six of the 38 patients in their series were treated with stereotactic radiosurgery for recurrence, with age at time of initial presentation ranging between 1 and 6 years. At the time of initial disease presentation, all patients received radiation therapy while three received chemotherapy. Notably, only two of the patients treated with SRS had prior gross total resection. Using a median dose of 18 Gy, ranging between 15 and 20 Gy, only one of the six patients survived. Four patients had local progression, though two of these patients had a longer PFS after SRS than after initial adjuvant therapy. This finding may suggest that SRS is better for temporizing local disease progression than standard radiotherapy when repeat surgery is not feasible. An even more encouraging result came from Liu et al. [75], who used hypofractionated re-irradiation, “staged radiosurgery,” to treat locally recurrent ependymomas in six children. All patients survived with no evidence of disease at median follow-up of 28 months from the time of re-irradiation (range 6–56 months), though it is important to note that five of the six patients had GTR upon disease recurrence prior to re-irradiation. Following re-irradiation, three of the six patients were reported to have a longer second disease-free interval than the time to initial progression. In both the Merchant and Liu series, however, evidence of radiation necrosis was described in a few patients and thus remains a risk in radiosurgical treatment (Case Study 31.2, Figs. 31.5 and 31.6).
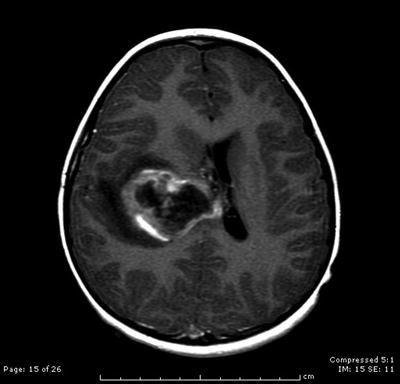
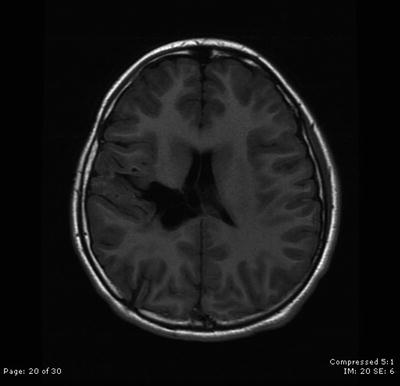
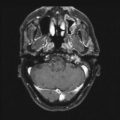
Fig. 31.5
Axial contrast-enhanced T1-weighted MRI scan showing a large right calcified and hemorrhagic paraventricular mass, diagnosed after biopsy as a supratentorial ependymoma
Fig. 31.6
The patient remains stable with no evidence of disease 5 years after surgical resection followed by intensity-modulated radiation therapy and concomitant chemotherapy
PNET (Medulloblastoma, Ependymoblastoma, Pineoblastoma)
Although there is some controversy surrounding the histopathological classification of childhood CNS embryonal tumors such as CNS primitive neuroectodermal tumors (PNETs), medulloblastoma, ependymoblastoma, and pineoblastoma (usually grouped with tumors of the pineal region), determining the appropriate surgical management plan and treatment is similar. Dissemination at the time of diagnosis for medulloblastomas and pineoblastomas to other intracranial sites and spinal cord is reported to occur in 10–30 % of patients and results in a worse prognosis [76–81]. CNS PNETs account for 4.8 % of childhood brain tumors [82]. Even with multimodal treatment, including maximal safe resection, fractionated neuraxis radiation therapy, and systemic chemotherapy, local and metastatic recurrences occur in 30–40 % of pediatric patients [83]. Five-year survival for standard-risk and high-risk patients are 70–80 % and 55–76 %, respectively [84]. Gamma knife surgery (GKS) has been found in anecdotal case series to have potential benefits especially for young children unsuitable for initial or additional fractionated radiation therapy, which carries the risk of neurotoxicity and poor neurocognitive outcome [85, 86]. Flannery et al. [87] showed that patients who had a smaller tumor volume were more likely to respond to GKS treatment. The longest survival, 126 and 67 months, occurred in two patients who had repeat maximal resection followed by early GKS.
Medulloblastoma
Medulloblastomas account for 20 % of pediatric CNS tumors and 40 % of posterior fossa tumors in children, with an incidence of 0.6 per 100,000 cases [88]. These tumors are most commonly found in fourth ventricle and tend to invade normal cerebellar tissue, with 15 % of cases infiltrating the brain stem [89]. Medulloblastomas are often seen as a midline hyperdense cerebellar vermian mass upon review on a CT scan [90], and ventriculomegaly is noted in approximately 85–90 % of all cases [91, 92]. Though the peak incidence of children diagnosed with medulloblastomas is between 5 and 7 years, 10 % of cases are diagnosed in the first year of life [93]. Patients with these tumors can be divided into standard risk and high risk. Standard risk patients are those over the age of 3, have no evidence of neuraxis dissemination on the brain and spine MRI and lumbar CSF, and have less than 1.5 cm2 of residual tumor on post-MRI scans. In contrast, high-risk patients are those with disseminated disease, anaplastic histology, postoperative CSF cytology, and a post-op MRI showing greater than 1.5 cm2 residual tumor [93, 94]. For children with medulloblastoma, surgery is usually the primary indication for treatment to remove as much of the tumor as safely possible while also confirming histological diagnosis. Maximal surgical resection has been associated with improved rate of survival, chiefly in those with no evidence of dissemination at the time of diagnosis [95, 96]. In infants, treatment is particularly challenging due to the vascular nature of tumor, high incidence of leptomeningeal spread, and lower chance of achieving a gross total resection [93]. Patrice et al. [97] was the first to propose the SRS technique as an effective and safe treatment in the management of medulloblastoma, showing that SRS was capable of controlling small, locally recurrent disease in 5 out of 11 patients with a median survival from the time of SRS of 10 (range 5 to 59+) months. The six remaining patients died of progressive disease. Furthermore, this series proposed that SRS was an effective addition to craniospinal irradiation for patients with newly diagnosed medulloblastoma, as all three patients were alive at last follow-up without evidence of disease [97].
Ependymoblastoma
Ependymoblastomas are considered a subgroup of CNS PNETs, most frequently located in the supratentorial region, but may also arise in the infratentorial or spinal sites [98]. These tumors are classified as WHO grade IV due to their aggressive behavior with rapid growth and proclivity to disseminate into the craniospinal region [99]. Gerber et al. [99] reported in their series of 10 patients a median OS survival of 12 months, with longer survival of those with radiotherapy or chemotherapy. If intraventricular chemotherapy was part of treatment, a subcutaneous reservoir with an intraventricular catheter was implanted in the anterior horn of lateral ventricle. In this series, 5 year PFS and OS were 36.4 ± 14.5 % and 30.3 ± 15.9 %, respectively, though all patients eventually died of tumor progression [99].
Pineoblastoma
Pineoblastomas are pineal region tumors that can metastasize along the neuroaxis and may be locally invasive, spreading outside the pineal region through the subarachnoid space [100]. These tumors are relatively rare, accounting for 0.6 % of all childhood brain tumors [82]. The standard treatment includes maximal surgical resection followed by adjuvant cranial-spinal irradiation and systemic chemotherapy [101]. A literature review of patients with pineoblastoma revealed a worse prognosis for children aged ≤5 years compared with older children; these patients had a 5-year survival rate of 15 % and 57 %, respectively (p < 0.00001). Among all patients, 5-year survival rate increased with degrees of resection: 29 % for debulking, 53 % for STR, and 84 % for GTR [100]. For children under the age of 3 years, the prognosis is even worse, with a survival rate at 1 year of 0 % [102]. Radiotherapy may have a significant impact on survival. A series by Hinkes et al. [102] indicated that six older children (median 6.75 y, range 3.6–16.9 y) who underwent radiotherapy in a maintenance chemotherapy regimen brought all those who had not been in complete remission into remission and kept all others in remission with no evidence of remaining tumor (Case Study 31.3, Figs. 31.7 and 31.8).
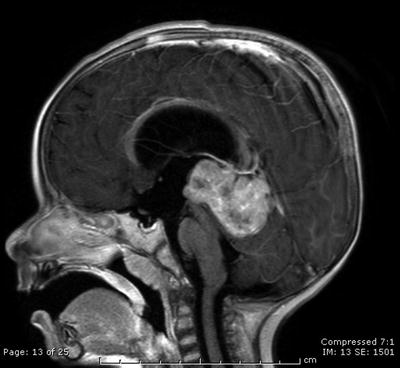
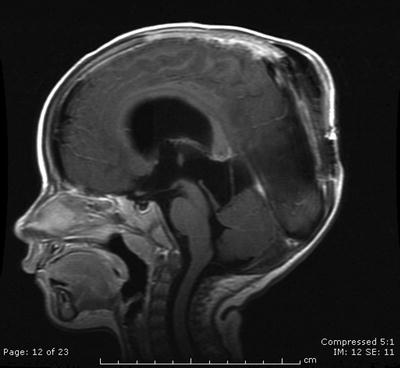
Fig. 31.7
A sagittal contrast-enhanced T1-weighted image reveals a large preoperative pineoblastoma in the pineal region
Fig. 31.8
The patient remains without residual tumor following surgical resection and chemotherapy at last follow-up
Vascular Malformations: Arteriovenous Malformation
The most dangerous congenital vascular malformations are arteriovenous malformations (AVMs). Prevalence in adults is about 18 per 100,000 and AVMs account for between 1 and 2 % of all strokes, 3 % of strokes in young adults, and 9 % of subarachnoid hemorrhages [103]. AVMs usually present between ages 10 and 40 with the most common symptoms being headache, seizures, focal neurologic deficits, and intracranial hemorrhage; however, at least 15 % of people affected by AVMs are asymptomatic [103]. The long-term crude annual case fatality rates are between 1 and 1.5 % [103]. After a hemorrhage, the annual mortality can be as high as 18 % [104]. Ninety percent of AVMs in the brain are located supratentorially, while the rest are located in the posterior fossa. The Spetzler-Martin Intracranial AVM grading scale [105] was devised based upon size, location, and deep venous drainage. The lesion grade (I–V) is derived by summing assigned points in each category. Complete surgical excision of a grade I lesion (small <3 cm, located in a non-eloquent region such as the anterior frontal lobe, and with solely superficial drainage) would have very little risk of any resulting morbidity or mortality. On the other hand, a grade V lesion (larger than 6 cm, located within or adjacent to eloquent brain areas and with some drainage into the deep venous system) would have significant morbidity and mortality [105]. Surgical resection is the treatment of choice for grade I–III AVMs, due to its high cure rate (reported from 89 to 98.4 %) and low morbidity and mortality [106, 107]. In children, AVMs are most frequently located in the basal ganglia, thalamus, corpus callosum, brainstem, or within the motor, speech, or visual cortex [108, 109] and are the most frequent cause of intracranial hemorrhage in children [110–112]. Mortality from AVM hemorrhage in the pediatric population is much greater for cerebellar AVM than for AVM in the cerebral hemisphere (57 % to 4.5 %, respectively, p < 0.0001) [113]. While surgery is the mainstay of treatment for AVMs, endovascular embolization has become a useful additional technique along with radiosurgery as an alternative for high-risk lesions. After primary stereotactic radiosurgery, Pan et al. [114] reported a pediatric AVM obliteration rate of 65 % at 48 months following the procedure. When needed, additional SRS led to a total obliteration rate of 81 % [114]. Kano et al. [110] showed an even higher total obliteration with SRS of 70 % with at least 4 years of follow-up. Similar obliteration rates were obtained with Gamma knife radiosurgery, as Dinca et al. [115] reported 71.3 % for a one time treatment and 82.7 % overall. Thus, GKS is considered to be a valid active management option for pediatric AVM, though limitations include delayed effect with persistence of bleeding risk for 2–4 years posttreatment [115] (Case Study 31.4, Figs. 31.9 and 31.10).
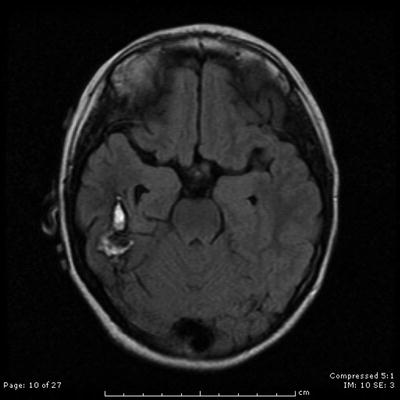
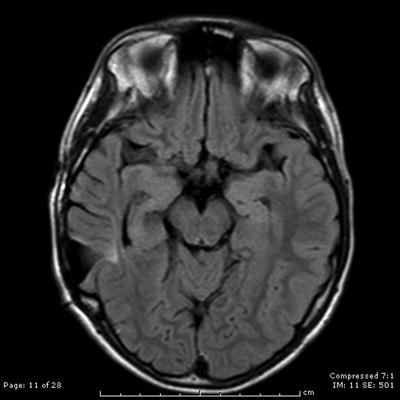
Fig. 31.9
An axial contrast-enhanced T2-weighted MRI shows the appearance of a right temporal lobe hematoma secondary to underlying AVM
Fig. 31.10
A year after recurrence and subsequent Cyber knife radiotherapy, an axial contrast-enhanced T2-weighted MRI scan shows no evidence of residual AVM
Case Studies
Case 31.1
The patient is a 15-year-old young man, who presented with an indolent history of progressive visual loss and headaches. Imaging studies revealed a large cystic craniopharyngioma (Fig. 31.1). A pterional craniotomy was then completed. Multiple cystic areas were resected and aspirated. This was a very prolonged and difficult dissection given the tumor’s investment in the surrounding optic chiasm, contralateral optic nerve, and posterior fossa. Two years later, the patient had evidence of tumor recurrence in the temporal lobe. Using microsurgical dissection techniques, the multiple cysts of the tumor were removed in this location. Two and a half years after the second resection, there was again evidence of recurrence, which required tumor removal in the medial portion of the sylvian fissure. Five months later, he underwent Gamma knife radiosurgery. The tumor remains controlled 1 year after radiosurgery at last follow-up (Fig. 31.2).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


