PET Radiopharmaceuticals: Fluorinated Compounds
Heinz H. Coenen
Dedicated to Professor Dr. Dr. H.C. Christoph Reiners on the occasion of his 60th birthday.
After short summaries of the basic features of fluorine-18 (18F) and 18F-labeling methods, a few selected radiofluorinated positron emission tomography (PET) tracers are briefly described. The focus is on clinically introduced 18F radiopharmaceuticals and those being presently established. It is clear that the most relevant fluorinated radiopharmaceutical is fluorodeoxyglucose (FDG), as amply documented in this book. It can now be produced consistently and reliably at 40 to 60 GBq (1 to 1.5 Ci) per batch and is being widely distributed; the only limitation for distribution is its short half-life of ∼110 minutes. However, additional fluorinated compounds are now available as potent diagnostic tracers for tumor imaging, such as 18FDOPA, 18FLT, and 18FET (Fig. 15.1), and for the diagnosis of receptor systems. Several selected new 18F-tracers with possible further diagnostic applications in the brain and heart and for tumor imaging are also discussed.
Introduction
The availability of radiopharmaceuticals labeled with fluorine-18 (18F) has made possible the widespread clinical use of PET. The chemical and nuclidic properties of 18F has led to a steady increase in the number of tracers concomitant with the increase in the variety of biochemical and pharmacological principles applied in molecular imaging. Various reviews have appeared recently describing the preparation of 18F-tracers for use in oncology and other diagnostic fields (1,2,3). Given the numerous tracers developed and the multiplicity of their applications, no exhaustive review can be given here. The focus will rather be on clinically used 18F-radiopharmaceuticals and those being established for this propose. Selected new 18F-tracers with the potential for use in further diagnostic areas are mentioned. Correspondingly, references can only be exemplary, under the aspect of availability, reflecting a critical consideration of labeling procedures developed for the individual compounds. The structures of most compounds mentioned are given in the figures.
The Radionuclide 18F As A Label
18F is the most important radionuclide for PET imaging besides carbon-11, and in the form of [18F]FDG it is the most often used in clinical studies. Whereas carbon-11 allows authentic labeling of biomolecules and pharmaceuticals, as well as repetitive studies (often for therapy control), because of its short half-life of 20.4 minutes, the decay properties of 18F provide different advantages. The low energy (635 keV) of the emitted β1-particle allows the highest spatial resolution among all PET radionuclides. Here the border of 1 mm
is attained with recently introduced small-animal PET scanners (4). The relatively longer half-life of 109.8 minutes offers the possibility of extended synthetic procedures and of following slower tracer kinetics of 6 hours or less. Thus, the time frame of study protocols is much less severe, with advantages for a clinical setting. Above all, 18F-labeled tracers can be provided to clinical services without a cyclotron, according to the so-called PET satellite concept.
is attained with recently introduced small-animal PET scanners (4). The relatively longer half-life of 109.8 minutes offers the possibility of extended synthetic procedures and of following slower tracer kinetics of 6 hours or less. Thus, the time frame of study protocols is much less severe, with advantages for a clinical setting. Above all, 18F-labeled tracers can be provided to clinical services without a cyclotron, according to the so-called PET satellite concept.
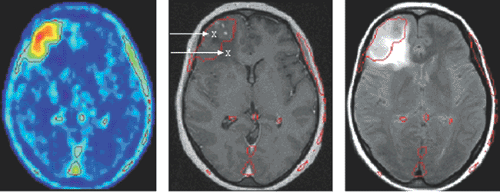 Figure 15.1 Uptake of O-(2-[18F]fluoroethyl)-L-tyrosine (FET) in a histologically verified oligodendroma III (left). The extent of abnormalities on the T2-weighted MRI (FLARE image on right) exceed FET accumulation in the PET scan, whereas those on the T1w contrast-enhanced scan (middle) are smaller in extension. Biopsy proved solid tumor only in the area of FET accumulation (43). (Courtesy of K.-J. Langen, Forschungszentrum Jülich.) |
Tagging a molecule with 18F in place of a hydrogen atom almost does not change its steric shape, and in general metabolically stable compounds are obtained. Its electronic influence, however, renders an analogous compound with changed physicochemical properties and with possibly altered biochemical, pharmacological, and toxicological features. This necessitates an evaluation of new 18F-labeled compounds with respect to their anticipated use as in vivo radiopharmaceuticals.
Principal Radiofluorination Methods
18F can be produced in good yields, even with modern low-energy medical cyclotrons (5). Quantities of greater than 150 GBq of 18F can be produced through the (p,n) nuclear reaction on enriched oxygen-18 (18O) at proton energies of less than 15 MeV. Most often [18O]water is used as target material, from which [18F]fluoride is obtained with a high specific activity of greater than 500 GBq/μmol. There is now no other practical way to produce the radionuclide in electrophilic form, besides adding elemental fluorine, but this limits the specific activity to less than 1 GBq/μmol. Thus, despite many new methodological improvements and various new pathways, the principal features of no-carrier-added (nca) radiofluorination, continuously reviewed over the last 20 years, are still valid (1,6,7,8,9).
Thus nucleophilic substitution with nca [18F]fluoride is still the only practical way to obtain labeled products with high specific activity. This is always necessary if a mass action of established radiopharmaceuticals or new tracers under development would lead to pharmacodynamic or toxic effects and hamper the tracer signal, especially in cases of low concentrations of the target molecules, as with receptors or enzymes (cf. ref. 10). For tagging 18F to aliphatic carbon, nucleophilic substitution is the method of choice, and various leaving groups for replacement are known for effective labeling. This also is true for activated aromatic derivatives (i.e. those that bear electron-withdrawing groups in addition to good leaving groups on the benzene ring). If electron-rich aromatic compounds have to be radiofluorinated on an nca level, electrophilic substitution is excluded, and products must be built up by multi-step synthetic procedures starting from a simple nca [18F]fluorobenzene derivative. This is generally tedious, makes automation difficult, and often hinders broad clinical use of a potent compound. For this purpose, however, many versatile nca 18F-labeled synthons have been developed (1,8,9).
As another alternative, prosthetic group labeling can often be used. In this method, a small functionalized nca [18F]fluoroalkyl or -aryl group is tagged to a target molecule of higher molecular mass. It is the method of choice for radiofluorination of oligomeric compounds like nucleotides (11) and especially peptides and proteins, as recently reviewed (12). It is also receiving increased interest as exemplified for markers of angiogenesis (13) and somatostatin receptors (14).
Established 18F- Radiopharmaceuticals
From the several hundred 18F-labeled tracers prepared for PET over the last 3 decades, only a few actually have reached the status of “established” 18F-radiopharmaceutical (Fig. 15.2). None has attained the importance and frequency of use of 2-[18F]fluoro-2-deoxy-D-glucose (18FDG), which is therefore referred to as the “work horse” of PET. Since its first proof as an indicator of energy metabolism (glucose consumption) (15), the number of indications for which it is a useful diagnostic compound have steadily increased, especially in oncology (16,17). Recently a complete compilation of the 18FDG-PET literature has been published (18).
A key prerequisite for the broad routine application of a radiopharmaceutical is its general availability (i.e. there must be a reliable radiochemical method of preparation). First developments of electrophilic labeling methods did not deliver 18FDG in high activity, and only a stereochemically impure product was available. Through the introduction of a nucleophilic nca radiofluorination on a tetraacetylmannose triflate precursor (19), the pure D-fluorodesoxyglucose became available in batch yields of 40 GBq or more. This method has been in use now for 20 years, with small improvements, especially a switch to basic hydrolysis of protecting groups (20,21). Thus 18FDG became the first PET radiopharmaceutical licensed in many countries. For a recent review on 18F-labeling of sugars, see Beuthien (22). The availability of this important diagnostic compound was and is the basis for the worldwide establishment of clinical PET.
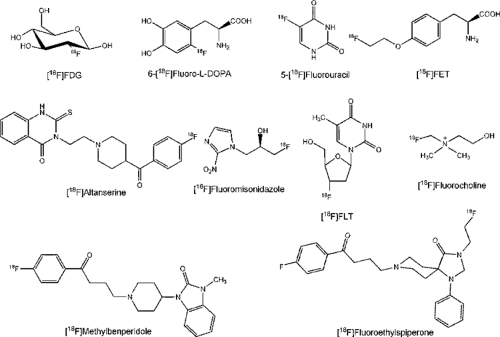 Figure 15.2 Established 18F-radiopharmaceuticals. |
The other established radiopharmaceuticals fall short in comparison with 18FDG because they are appropriate for a much more limited number of clinical indications and in many cases lack availability (i.e. ease of radiosynthesis). The latter shortcoming does not apply to nca [18F]fluoride, which only must be transferred upon its production in a [18O]water target to an isotonic injectable solution. The chemically most stable form of fluorine, its anion, represents (as [18F]NaF) a radiopharmaceutical for bone imaging, as suggested 30 years ago (23,24). After 18FDG and 6-[18F]fluoro-L-dopa (18FDOPA), it ranks third in frequency of clinical use and is specifically employed for detection of bone metastases.
In contrast to 18FDG, 18FDOPA and 5-[18F]fluorouracil (5-18FU) are generally still produced by electrophilic methods, with the disadvantage of relatively small batch yields and limited availability. The chemotherapeutic analog 5-FU is produced by an addition–elimination sequence of
electrophilic fluorine species on dihydrouracil (25). It is used for therapy control (26) and has been discussed as a means of predicting therapy response in liver metabolism in patients with colorectal carcinoma (27). Although nucleophilic procedures were established for the routine production of 18FDOPA, they are difficult to implement because of multi-step radiosyntheses. Most efforts were devoted to improve the carrier-added (ca) electrophilic radiofluorination. To date, a fluorodestannylation reaction (28) appears to be the best option for regioselective labeling with satisfactory yields. This allowed automation (29), and recent improvements in precursor preparation and quality control (30) ensure reliable production for in-house application.
electrophilic fluorine species on dihydrouracil (25). It is used for therapy control (26) and has been discussed as a means of predicting therapy response in liver metabolism in patients with colorectal carcinoma (27). Although nucleophilic procedures were established for the routine production of 18FDOPA, they are difficult to implement because of multi-step radiosyntheses. Most efforts were devoted to improve the carrier-added (ca) electrophilic radiofluorination. To date, a fluorodestannylation reaction (28) appears to be the best option for regioselective labeling with satisfactory yields. This allowed automation (29), and recent improvements in precursor preparation and quality control (30) ensure reliable production for in-house application.
After introduction of 18FDOPA (31), it became a well-established tracer for studying presynaptic integrity of dopaminergic innervation, mainly in Parkinson disease (32,33). Although the uptake of 18F-labeled DOPA in melanomas has been known for 15 years, it has only recently found applications in oncology, with great promise as complementary tracer to 18FDG (for review, see ref. 3). In neuroendocrine tumors, for example, it has been shown to be superior to somastotatin receptor scintigraphy (34). The report of a carcinoid crisis induced by 18FDOPA points to the necessity of a practical nca labeling method for this tracer (35).
Generally, tumor accumulation of amino acids is dominated by transport much more than by protein synthesis. Thus various attempts were aimed at 18F-labeled amino acid analogs specific for different transporter systems; for reviews on [18F]fluoroamino acids, see refs. 36 and 37. The most potent one appears to be O-(2-[18F]fluoroethyl)-L-tyrosine (FET), which was first prepared using a two-step fluoroalkylation method (38) but can now be produced using direct substitution, one-pot synthesis, with high batch yields like 18FDG for broader clinical use (39). Besides the logistic advantages, it was found not to accumulate in inflammatory tissue like 18FDG (40) or methionine (41), although it shares a similar amino acid transporter profile. Above all, biopsy samples of human gliomas have proved that in cases of FET-PET and MRI mismatch the amino acid indicated the solid tumor with much higher specificity (42) (see Fig. 15.1). Meanwhile, it has led to convincing results, especially in the diagnosis of brain tumors, suggesting perspectives as a clinical standard tracer (43).
Based on the findings from imaging prostate cancer with [11C]choline, [18F]fluorocholine was prepared and shown in initial patient studies to detect prostate cancer and metastases thereof clearly (44). Because the radiosynthesis involved intermediate gas chromatographic purification steps, the [18F]fluoroethyl analog was also prepared. Although poorer biologic compatibility was found in in vitro cell experiments (45), it proved very effective for detecting prostate cancer with PET (46). The biodistribution of both compounds is very similar to that of [11C]choline, but their urinary excretion is much more rapid, demanding appropriate imaging protocols (Chapter 49) or bladder catheterization (45). [18F]fluorocholine seems advantageous and is also detected in intracranial lesions (47).
Another very promising tracer for tumor imaging is 3-deoxy-3′-[18F]fluorothymidine (18FLT), as it is an indicator of cell proliferation (48), is stable in vivo, and is a false substrate of thymidine kinase. Whereas the first nucleophilic nca synthesis starting from an anhydro precursor had low radiochemical yields, a new precursor allows better and reliable radiosynthetic results, thus improving clinical availability (49). Accumulation of the compound in normal proliferating tissue is a drawback with respect to bone marrow dosimetry, as this compound accumulates strongly in the hematopoietic bone marrow. Conversely, FLT scans may thus permit control of the action of chemotherapy on critical organs. Based on recent findings, this radiotracer shows clinical suitability (2,50,51) in comparison with 18FDG (52) and [11C]thymidin (53).
A high specific activity of the radiopharmaceutical is an absolute necessity for imaging of receptors because of their low concentration (see ref. 10). This requirement has been met for several radioligands of central and peripheral neurotransmission systems by using nca nucleophilic substitution procedures. Examples for the 5-HT2A serotonin receptor are [18F]setoperone and [18F]altanserin, with the latter more widely accepted (54,55,56). Both ligands are prepared from the nitro-substituted precursor, with good radiochemical yields (54,57,58). A database on the binding of [18F]altanserin to 5-HT2A receptors was recently constructed using normal volunteers (59).
Related posts:
 11C-Based PET- Radiopharmaceuticals of Clinical Value
11C-Based PET- Radiopharmaceuticals of Clinical Value
 Brain Tumors: Astrocytoma, Oligodendroglioma, and Glioblastoma Images with PET and SPECT
Brain Tumors: Astrocytoma, Oligodendroglioma, and Glioblastoma Images with PET and SPECT
 Sentinel Node Imaging in Head and Neck Tumors
Sentinel Node Imaging in Head and Neck Tumors
 PET-CT and SPECT-CT of Bone Metastases
PET-CT and SPECT-CT of Bone Metastases
 SPECT and PET in Benign Thyroid and Parathyroid Disease
SPECT and PET in Benign Thyroid and Parathyroid Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


