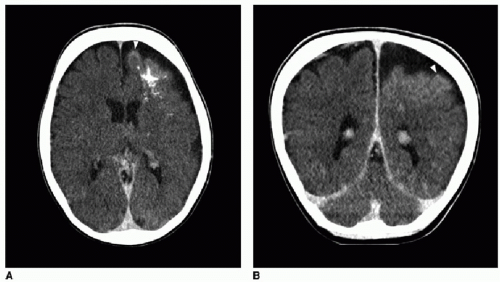
FIG. 32.2 • Examples of MRI findings seen in Sturge-Weber syndrome (SWS). A: Axial T1-weighted fluid-attenuation inversion recovery (FLAIR) image of a 1-day-old neonate already shows mild left cerebral volume loss and advanced myelination, evidenced by asymmetrically increased T1 hyperintensity in the left-sided white matter (
arrows).
B: Axial susceptibility-weighted image of the same neonate shows increased number of hypointense veins within the extra-axial space (
white arrows), consistent with leptomeningeal angiomatosis. Linear dark signal through the left frontal lobe (
black arrowhead) represents a transmedullary vein. Globular susceptibility artifact in the left frontal periventricular white matter (
black arrow) represents calcification.
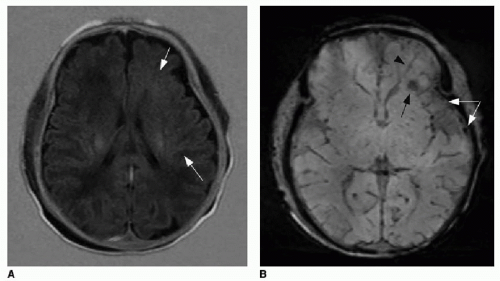
C: Axial T1-weighted image of the same patient imaged later at age 5 months shows area in the superior medial left frontal lobe with abnormal blurring of the gray-white matter junction (
arrow). Postcontrast T1-weighted
(D) images of superior left hemisphere and at level of inferior
(E) left frontal lobe show abnormal cortical enhancement (
arrows).
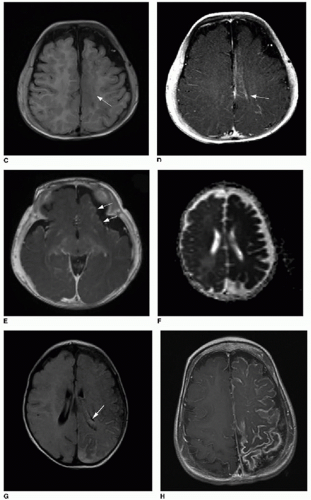
F: Apparent diffusion coefficient (ADC) map reconstructed from diffusion-weighted imaging of different patient at 6 weeks of age, showing diffuse left cerebral hemisphere dark signal, indicating restricted diffusion and ischemia.
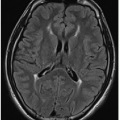
G: Axial T2-weighted FLAIR image of same study in a 6-week-old infant shows extensive left-sided subarachnoid hyperintense signal. Note also the asymmetrically larger left choroid plexus (
arrow), typical of SWS.
H: Axial T1-weighted postcontrast image of a 2-year-old patient (same patient as shown in
Fig. 32.1) shows extensive, abnormal leptomeningeal and gyriform enhancement along the left cerebral hemisphere.