CHAPTER 99 Pulmonary Hypertension
PULMONARY HYPERTENSION
Definition
Pulmonary hypertension (PH) is defined as a resting mean pulmonary arterial pressure more than 25 mm Hg at rest or more than 30 mm Hg with exercise. Among the various subclassifications of PH, the subgroup referred to as pulmonary arterial hypertension adds the criterion that the pulmonary capillary wedge pressure should be 15 mm Hg or less.2
Etiology and Pathophysiology
Anatomy and Physiology of the Pulmonary Circulation
The pulmonary circulation consists of two parallel networks, the pulmonary arterial circulation and bronchial arterial circulation.1 The pulmonary arterial circulation consists of a series of branching arteries, arterioles, capillaries, venules, and veins. Pulmonary arteries travel adjacent to the lobar, segmental, and subsegmental airways to the level of the terminal bronchioles. The small pulmonary arteries from the subsegmental level to the terminal bronchioles possess a thick muscular media. At the level of the respiratory bronchioles and alveolar ducts, the small pulmonary arteries have lost much of the muscle within the arteriolar media as well as their external elastic membranes, and are now termed pulmonary arterioles, ranging in size from 10 to 150 µm. These vessels ramify further within the alveolar walls and form a rich capillary network. Capillary blood collects in venules, which progressively coalesce to form pulmonary veins. Pulmonary veins course within interlobular septa, eventually emptying into the left atrium.
Pathogenesis
The pulmonary vascular endothelium actively responds to changes in oxygen tension, transmural pressure, and pulmonary blood flow, and actively participates in the regulation of pulmonary arterial pressure through the elaboration of a number of vasoactive substances, including prostacyclin, nitrous oxide, and endothelin.3 These agents have direct effects on pulmonary vascular smooth muscle tone, promoting relaxation or vasodilation, and also may directly affect platelet function.
Various pathologic derangements may be seen in patients with PH, varying somewhat depending on the cause of PH. In general, regardless of the cause of PH, the pulmonary arteries become dilated,1 occasionally to the point of being considered aneurysmal. Pulmonary arterial atherosclerosis, typically absent in the pulmonary arteries of normal adults, may be extensive and frequently involves smaller vessels in patients with PH. PH-related pulmonary arterial atherosclerosis histopathologically appears similar to atherosclerosis occurring in systemic arteries, although complicating features, such as necrosis, ulceration, and calcification, are relatively uncommon.
Plexogenic pulmonary arteriopathy is the term applied to a constellation of vascular changes often encountered in patients with primary PH, but may also be seen in patients with PH of other causes, including hepatic disease, connective tissue disorders, congenital cardiovascular disease, and some anorexigenic medications.2
Classification
There have been a number of proposed classification schemes to categorize the causes of PH. In 1998, during the Second World Symposium on Pulmonary Hypertension held in Evian, France, a clinical classification for PH was proposed (Box 99-1).3–5 The aim of the Evian classification was to provide categories for causes of PH that shared similar pathophysiologic mechanisms, clinical presentations, and therapeutic considerations. Similar to oncologic staging considerations, such a classification was designed to allow standardization of diagnosis and treatment for patients with PH, conduction of trials with similar patient cohorts, and investigation of pathobiologic abnormalities in patient populations that are highly characterized. In 2003, the Third World Symposium on Pulmonary Arterial Hypertension, held in Venice, evaluated the usefulness of the Evian classification system and recommended some minor modifications. The Venice classification was adopted by the World Health Organization (WHO) and has been referred to as the Revised World Health Organization Classification of Pulmonary Hypertension (Box 99-2). More recently, the revised WHO-Venice classification underwent revision at the Fourth World Symposium on Pulmonary Hypertension held in Dana Point, California. The new classification system is referred to as the 2008 Dana Point Pulmonary Hypertension classification system.6 Traditionally, pulmonary hypertension has been classified into primary versus secondary causes or precapillary and postcapillary causes; this classification scheme is still in common use, particularly among interpreting physicians, and is included here as well. The pre- and postcapillary PH classification scheme is commonly reviewed in radiology texts (Box 99-3),1 but the 2008 Dana Point classification system is in more widespread use among clinicians and investigators dealing with PH. The 2008 Dana Point classification system will be used for this chapter.
BOX 99-1 Classification of Pulmonary Hypertension: Evian Classification
BOX 99-2 Classification of Pulmonary Hypertension: Venice-Revised WHO Classification
I PULMONARY ARTERIAL HYPERTENSION
BOX 99-3 Classification of Pulmonary Hypertension: Precapillary Versus Postcapillary Etiologies








In the 2008 Dana Point classification schemes, the category of pulmonary arterial hypertension (PAH) includes a subgroup of patients with PH without any identifiable cause, or idiopathic PAH,6 previously referred to as primary pulmonary hypertension (PPH). Other subgroups in the category of PAH include disorders that share localization of the lesions to small, muscular pulmonary arterioles, including heritable causes of PH (e.g., patients with mutations in bone morphogenetic protein receptor 2 and activin receptor-like kinase 1), drug and toxin exposures, and associations with certain conditions, such as collagen vascular diseases, HIV-related PH, portopulmonary hypertension, congenital systemic to pulmonary artery shunts, schistosomiasis, and chronic hemolytic anemias. The morphologic and clinical characteristics of these disorders are similar, and they also share a clinical response to treatment with continuous infusion of epoprostenol.5 Most patients previously diagnosed with primary pulmonary hypertension would now be classified as patients with idiopathic pulmonary arterial hypertension (IPAH) or, less commonly, heritable PH or anorexigen-induced PH (Box 99-4).7 Persistent PH of the newborn was also recognized as a cause of PAH in the 2008 Dana Point PH classification system. The Venice-revised WHO classification system also includes another subcategory in the pulmonary arterial hypertension group, referred to as “pulmonary arterial hypertension associated with significant venous or capillary involvement.” This subcategory includes pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. In the 2008 Dana Point system, this subcategory remains a group 1 (PAH) condition, but is now referred to as “pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis” in recognition of the similarities between these two disorders (see Box 99-4). Both entities were classified differently in the Evian system.
BOX 99-4 Classification of Pulmonary Hypertension: 2008 Dana Point Classification
The category referred to as pulmonary venous hypertension in the Evian system and as PH with left heart disease in the Venice-revised WHO system is now called pulmonary hypertension secondary to left heart disease (see Box 99-4). It includes systolic and diastolic dysfunction as well as left-sided valvular and myocardial conditions that require therapies directed at improving cardiac function or repairing or reducing valvular mechanical dysfunction, as opposed to treatment with vasodilator therapy.5,6 In fact, epoprostenol infusion in these patients can be harmful. Pulmonary veno-occlusive disease and lesions producing extrinsic compression on pulmonary veins are included in this category in the Evian system, but were reclassified in the Venice-revised WHO system in 2003 and again in the 2008 Dana Point system.
PH secondary to hypoxia and/or lung disease (see Box 99-4) results from inadequate arterial blood oxygenation caused by chronic obstructive pulmonary disease, interstitial lung diseases, sleep-disordered breathing, alveolar hypoventilation, and residence at high altitude. Usually, the elevated pulmonary arterial pressures in these patients are fairly modest (<35 mm Hg).
Group 4 in the 2008 Dana Point PH classification system is chronic thromboembolic pulmonary hypertension (see Box 99-4).6 In many patients with chronic thromboembolic PH, the central pulmonary arteries show emboli and/or thrombi, and such patients may benefit from pulmonary endarterectomy.1 In other patients, emboli or thrombi are relatively distally located and are not amenable to surgical treatment. These patients may respond to chronic pulmonary vasodilator therapy, similar to patients with IPAH. Both subgroups of patients are treated with lifelong anticoagulation.
PH caused by disorders of the pulmonary vasculature in the Evian system was simply referred to as the miscellaneous category in the Venice-revised WHO system, and is now called pulmonary hypertension with unclear multifactorial mechanisms in the 2008 Dana Point classification. Included in this group are a variety of disorders such as myeloproliferative diseases, splenectomy, cystic lung diseases, sarcoidosis, and metabolic disorders. External vascular compression, particularly fibrosing mediastinitis, previously considered a subcategory of pulmonary arterial hypertension in the Evian system, was moved to the miscellaneous category in the Venice-revised WHO system and is now classified as a group 5 lesion in the 2008 Dana Point system.6
Manifestations
Imaging Techniques and Findings
The characteristic finding of pulmonary arterial hypertension on chest radiography, CT, or MRI is dilation of the central pulmonary arteries, with rapid tapering of the pulmonary vessels as they course peripherally.1 This pattern is present regardless of the cause of the PH.
Radiography
Chest radiography may show enlargement of the main pulmonary artery segment and dilation of the right and left interlobar pulmonary arteries in patients with PH, regardless of cause (Fig. 99-1).
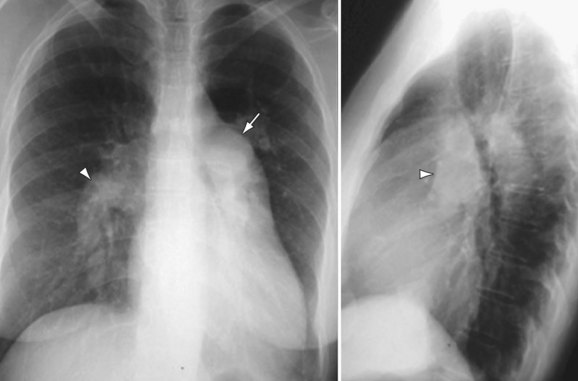
 FIGURE 99-1
FIGURE 99-1Computed Tomography
The main pulmonary arterial segment cannot be directly measured on chest radiography, but is easily measured on CT or MRI. When the main pulmonary artery segment exceeds 3 cm (Fig. 99-2), PH is often present. However, PH may be present in patients with a normal-sized main pulmonary arterial segment. The main pulmonary artery–to–ascending aortic ratio is also a useful internal indicator that can suggest enlargement of the main pulmonary artery. When the ratio of the main pulmonary artery to the aorta exceeds 1, elevated pulmonary pressures are usually present.1
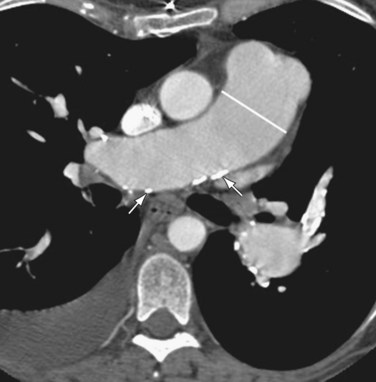
 FIGURE 99-2
FIGURE 99-2When PH is prolonged and severe, calcification of the pulmonary arteries, usually affecting the main, right, and/or left pulmonary arteries and, less commonly, the lobar pulmonary arteries, may be present (see Fig. 99-2). This finding is usually, but not invariably, associated with irreversible pulmonary vascular disease.
Patients with elevated pulmonary arterial pressures may also show right ventricular enlargement and right ventricular hypertrophy (Fig. 99-3) on cross-sectional imaging studies.1 The right atrium and inferior vena cava may also be enlarged, and contrast reflux into the infradiaphragmatic inferior vena cava is also commonly seen.
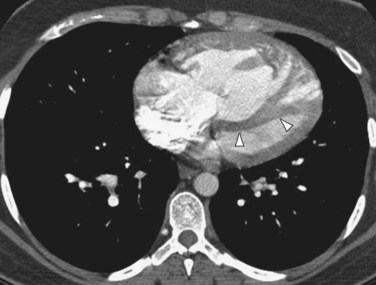
 FIGURE 99-3
FIGURE 99-3Magnetic Resonance Imaging
MRI will show all the vascular abnormalities that thoracic CT detects in patients with PH, such as main pulmonary artery enlargement and right ventricular enlargement and hypertrophy.1 Additionally, MRI can provide functional information equivalent to echocardiography, such as direction and velocity of blood flow, in addition to specific anatomic information. MRI techniques are well suited to the evaluation of patients with PH because they allow a detailed anatomic and extensive functional examination of the entire cardiovascular system.
IDIOPATHIC PULMONARY ARTERIAL HYPERTENSION
Definition
The term primary pulmonary hypertension was first used in 1951 to describe hypertensive vasculopathy in the pulmonary arteries without an identifiable cause.5 More recently, it has been recognized that patients with heritable forms of PAH and those with various conditions associated with PH, including connective tissue diseases, the use of appetite suppressant medications, portopulmonary hypertension, and HIV infection, among others, may share pathologic and clinical features similar to PPH, and were previously collectively referred to as secondary PH. The disorders encompassed by the term secondary PH are a fairly heterogeneous group of conditions that also include conditions affecting the pulmonary venous circulation or pulmonary parenchyma as well. The use of secondary PH has been largely abandoned because it does not provide a useful framework for the diagnosis or treatment of patients with PH. Therefore, the Venice group recommended changing the term primary PH to idiopathic pulmonary arterial hypertension; this nomenclature was adopted in the Venice-revised WHO classification system and is maintained in the 2008 Dana Point classification system.
Etiology and Pathophysiology
The cause of IPAH is unknown. The prevalence of PAH is estimated at about 15/million.7 The disease affects females more commonly than males. The Venice-revised WHO classification system previously recognized two forms of IPAH, familial and idiopathic (sporadic). The 2008 Dana Point classification system now refers to familial PAH as heritable PAH, and considers IPAH as a separate subcategory of PAH (see Box 99-4).6 Heritable forms of PAH account for approximately 10% of PAH cases, show autosomal dominant inheritance with incomplete penetrance, and have been localized to chromosome 2 at the locus of the bone morphogenic protein receptor type II gene.5 Heritable PAH associated with mutations in activin receptor-like kinase type 1 may be associated with hereditary hemorrhagic telangiectasia.
Manifestations of Disease
Clinical Presentation
Patients with IPAH usually present with dyspnea on exertion. The mean age of diagnosis is approximately 37 years.7 Other presenting symptoms include fatigue, chest pain, syncope, and occasionally cough.
Imaging Techniques and Findings
Radiography
Chest radiography in patients with IPAH typically shows enlargement of the main, right, and left pulmonary arteries (see Fig. 99-1), often with enlargement of the right ventricle (see Fig. 99-3) and right atrium.1 CT and black blood MRI techniques will show these same findings to advantage. Occasionally, black blood MRI will show increased signal within the pulmonary arteries resulting from slow flow.
Computed Tomography
High-resolution CT (HRCT) in patients with IPAH may show inhomogeneous lung opacity resulting from differential pulmonary parenchymal perfusion (Fig. 99-4).1 The regions of decreased pulmonary parenchymal attenuation represent areas of mosaic perfusion, and the vessels in these regions of lung may be visibly smaller than their counterparts in the regions of relatively increased lung attenuation. Although airway diseases may result in a similar pattern of mosaic perfusion, vascular and airway causes of mosaic perfusion may be distinguished using postexpiratory imaging. When caused by an obstructive airway process, regional differences in lung attenuation become accentuated with postexpiratory imaging, whereas a proportional increase in attenuation in areas of both increased and decreased attenuation will be seen in patients with pulmonary vascular disease.
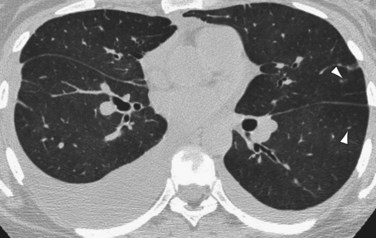
 FIGURE 99-4
FIGURE 99-4Occasionally, HRCT will show centrilobular ground-glass opacities in patients with IPAH (Fig. 99-5), representing foci of hemorrhage, plexiform lesions, or cholesterol granulomas.8
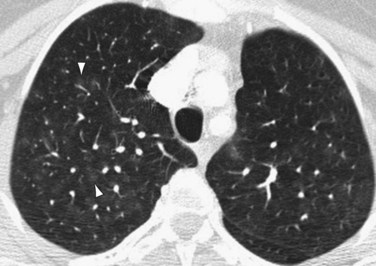
 FIGURE 99-5
FIGURE 99-5Nuclear Medicine and Positron Emission Tomography
Ventilation-perfusion (V/Q) scintigraphy is commonly abnormal in patients with IPAH, but the findings are usually nonspecific and are often interpreted as representing a low probability for pulmonary thromboembolic disease.1
PULMONARY ARTERIAL HYPERTENSION RELATED TO RISK FACTORS OR ASSOCIATED WITH CONDITIONS
Definition
Pulmonary arterial hypertension related to associated conditions (APAH) includes PH in patients with connective tissue diseases, HIV infection, portal hypertension, congenital cardiovascular systemic to pulmonary shunts, schistosomiasis, and chronic hemolytic anemias.6 The term risk factors in the category of PAH indicates any condition suspected to play a role in predisposing or facilitating the development of PAH, whereas the term associated conditions is used when it is impossible to determine whether a predisposing factor was present prior to the development of PAH.5 The Evian conference in 1998 explored a number of potential risk factors and classified them according to the strength of association with PAH and the likelihood of a causal role in the development of PAH as definite, very likely, possible, or unlikely, according to the strength of available evidence in the literature (Box 99-5).
Etiology and Pathophysiology
PAH associated with connective tissue disease usually occurs in patients with systemic sclerosis, particularly those with limited forms of this disorder, such as CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).7,9 Although most patients with systemic sclerosis do not develop clinical evidence of PAH, histopathologic changes of PAH are common in patients with systemic sclerosis. Echocardiographic studies have suggested that mild or moderate PAH is not uncommon in patients with systemic sclerosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


