Many different pulmonary manifestations are seen in conjunction with genetic disorders. Pulmonary findings have been noted with some cytogenetic conditions, many single gene or mendelian disorders, and numerous inborn errors of metabolism. In addition, congenital lung anomalies are common, occurring as isolated anomalies and as part of multiple anomaly syndromes. Recognition of pulmonary problems in patients with genetic disorders may lead to prompt treatment and intervention, which ultimately might translate into improved outcome.
Congenital Anomalies
Overview of Prenatal Lung Development
Lung morphogenesis can be subdivided into distinct periods on the basis of the morphologic characteristics of the tissue ( Table 14-1 ).
| PERIOD | GESTATIONAL AGE (WK) | STRUCTURAL EVENTS |
|---|---|---|
| Embryonic | 3–6 | Lung buds, trachea, main stem, lobar, and segmental bronchi |
| Pseudoglandular | 6–16 | Subsegmental bronchi, terminal bronchioles, acinar tubules, mucous glands, cartilage, smooth muscle |
| Canalicular | 16–26 | Respiratory bronchioles, acinus formation and vascularization, type I and type II cell differentiation |
| Saccular and alveolar | 26–36 (saccular); 36 to maturity (alveolar) | Dilation and subdivision of alveolar saccules, increase of gas-exchange surface area, further growth and alveolarization, maturation of alveolar-capillary network |
Embryonic Period
Prenatal lung development begins during the embryonic stage of fetal development, at 4 weeks gestation, with lung buds emerging from the ventral side of the foregut, which is lined by endodermally derived epithelium. The trachea is separated from the primitive esophagus beginning at week 5. Secondary bronchi then form, three on the right and two on the left, which ultimately form the five lobes of the lungs. Developmental anomalies occurring during this period of development may include tracheal, laryngeal, and esophageal atresia; tracheal stenosis; pulmonary agenesis; tracheoesophageal fistulas (TEFs); and bronchial malformations.
Pseudoglandular Period
The pseudoglandular stage is so called because of the distinct glandular appearance of the lung from 6 to 16 weeks of gestation. During this period, branching of the airways continues, and formation of the terminal bronchioles and primitive acinar structures is completed by the end of this period. Surfactant proteins A, B, and C are first detected during this stage. Bronchial arteries arise from the aorta and form along the epithelial tubules. Various congenital defects may arise during this stage of lung development, including tracheobronchomalacia, pulmonary sequestration, cystic adenomatoid malformation, ectopic lobes, cyst formation, and congenital pulmonary lymphangiectasia. The pleuroperitoneal canal also closes early in the pseudoglandular period. Failure to close the pleural cavity, often accompanied by herniation of the abdominal contents into the chest (congenital diaphragmatic hernia [CDH]), leads to lung hypoplasia.
Canalicular Period
The canalicular stage occurs at 16 to 26 weeks of gestation. It is characterized by the formation of acinar structures in the distal tubules, thinning of the mesenchyme, and formation of the capillary bed. By the end of this period, the terminal bronchioles have divided to form two or more respiratory bronchioles, and each of these has divided into multiple acinar tubules, forming the primitive alveolar ducts and pulmonary acini. At the end of this stage in infants, gas exchange can be supported after birth. Abnormalities of lung development occurring during this period include pulmonary hypoplasia (caused by diaphragmatic hernia or compression by thoracic or abdominal masses and prolonged rupture of membranes causing oligohydramnios) and renal agenesis, in which amniotic fluid production is impaired.
Saccular and Alveolar Periods
Saccular and alveolar periods occur at 26 to 36 weeks and 36 weeks through adolescence. Increased thinning of the respiratory epithelium and pulmonary mesenchyme, further growth of the lung acini, and development of the distal capillary network characterize these stages. In the periphery of the acinus, maturation of type II epithelial cells occurs in association with increasing numbers of lamellar bodies and increased synthesis of surfactant phospholipids. Pulmonary arteries enlarge and elongate in close relationship to the increased growth of the lung. After birth, alveoli expand a bit, but, more importantly, lung growth remains active throughout the first 10 years of life. There is a sixfold increase in the number of alveoli during postnatal life.
Before birth, fetal respiratory movements allow lung fluid that is constantly produced to efflux into the amniotic fluid. These respiratory efforts promote further lung development and strengthen the muscles of respiration. As discussed in the next section, prenatal circumstances that prevent adequate respiratory movements before birth (i.e., oligohydramnios, thoracic cage abnormalities, or neuromuscular disease) may lead to life-threatening pulmonary hypoplasia.
Lung Agenesis and Pulmonary Hypoplasia
Primary pulmonary hypoplasia is occasionally reported as an isolated finding and has been described in many sibships, suggesting autosomal recessive inheritance in at least some cases. This serves as a reference point for some families who have given birth to one child with pulmonary hypoplasia because risks for recurrence may be 25% in subsequent pregnancies.
Tracheal atresia accompanied by bilateral agenesis of the lungs is most likely to arise as an isolated defect, possibly secondary to a vascular event occurring very early in the embryonic period. Variable degrees of pulmonary agenesis have been reported in many sibships, however, with and without consanguinity. Developmental anomalies of the lungs also have been described in association with other birth defects, as part of at least one possible mendelian disorder, and as an occasional finding in a microdeletion syndrome ( Table 14-2 ).
| SYNDROME | CLINICAL FEATURES | MODE OF INHERITANCE | GENE OR LOCUS IF KNOWN |
|---|---|---|---|
| Total anomalous pulmonary venous return (scimitar syndrome) | Right lung hypoplasia | Autosomal dominant with incomplete penetrance (40% penetrant) | Locus = 4q12 |
| Pulmonary hypertension | |||
| Total anomalous pulmonary venous return | |||
| Dextrocardia | |||
| Anophthalmia and pulmonary hypoplasia (Matthew-Wood syndrome) | Anophthalmia | Possible autosomal recessive | Unknown |
| Pulmonary hypoplasia (with or without diaphragmatic hernia) | |||
| Velocardiofacial syndrome (22q11.2 deletion syndrome) | Pierre-Robin sequence | Autosomal dominant with many new or de novo deletions | Locus = 22q11.2; cytogenetic microdeletion |
| Velopharyngeal insufficiency | |||
| Cardiac defects (especially septal or aortic arch) | |||
| Unilateral pulmonary dysgenesis | |||
| T cell defects | |||
| Hypocalcemia | |||
| Learning disabilities | |||
| Psychosis |
More often, pulmonary hypoplasia identified at birth in association with severe respiratory distress is found to be secondary to another underlying defect or disorder. These primary problems can be divided into three broad categories: (1) renal problems leading to oligohydramnios, (2) skeletal dysplasia resulting in thoracic cage limitation and pulmonary insufficiency, and (3) neuromuscular disorders leading to pulmonary hypoplasia owing to poor respiratory efforts in utero .
Any renal abnormality, developmental or obstructive, leading to decreased urine output in utero with concomitant oligohydramnios has the potential to cause pulmonary hypoplasia (see Chapter 6 ). The degree of oligohydramnios and length of time the fetus is exposed to limitation of movement likely determine the severity of pulmonary hypoplasia and potential for long-term survival. Most instances of oligohydramnios are not genetically determined, but instead are secondary to prolonged rupture of membranes. Late loss of amniotic fluid with a relatively short duration of fetal constriction is less likely to cause significant respiratory compromise. In contrast, prolonged, severe oligohydramnios secondary to renal agenesis or sirenomelia results in the Potter sequence with severe pulmonary hypoplasia and neonatal death.
In between these two extremes are variable findings and prognoses associated with decreased amniotic fluid volume. A common isolated birth defect in male fetuses, posterior urethral valves, may result in oligohydramnios, bladder distention, hydronephrosis, and ultimately renal damage. In severe cases, the bladder distention leads to anterior abdominal wall musculature hypoplasia and renal failure (prune-belly syndrome). These boys also may have respiratory compromise that tends to correlate with the duration and severity of the oligohydramnios.
Suboptimal urine output associated with many genetic conditions may be seen with varying degrees of pulmonary hypoplasia at birth. Table 14-3 lists some of these syndromes. Bilateral or unilateral renal agenesis may occur as an isolated (nonhereditary) defect during early fetal development.
| SYNDROME | CLINICAL FEATURES | MODE OF INHERITANCE | GENE OR LOCUS IF KNOWN |
|---|---|---|---|
| Hereditary renal agenesis | Pulmonary hypoplasia | Autosomal dominant (general recurrence risk of 1% in isolated cases—if parents have two normal kidneys) | Gene = PAX2 ; locus = 5q11.2-q11.3 |
| Renal agenesis | |||
| Other genitourinary anomalies | |||
| Potter facies | |||
| Secondary equinovarus | |||
| Autosomal recessive polycystic kidney disease (incidence 1:16,000 live-born infants) | Pulmonary hypoplasia | Autosomal recessive | Gene = PKHD1 |
| Enlarged cystic kidneys with interstitial fibrosis | |||
| Hepatic and pancreatic cysts | |||
| Potter facies | |||
| Meckel syndrome (Meckel-Gruber syndrome) | Pulmonary hypoplasia | Autosomal recessive | Gene = MKS1 on 17q; locus = 11q13 ( MKS2 ); 8q24 ( MKS3 ) |
| Polycystic kidneys with or without renal agenesis | |||
| Occipital encephalocele | |||
| Postaxial polydactyly | |||
| Nephronophthisis type 2 | Pulmonary hypoplasia | Autosomal recessive | Unknown |
| Hyperechogenic kidneys with cortical microcysts and tubulointerstitial nephritis | |||
| Genitopatellar syndrome | Pulmonary hypoplasia | Autosomal recessive | Unknown |
| Polyhydramnios (not oligohydramnios) | |||
| Microcephaly, CNS defects | |||
| Cardiac septal defects | |||
| Multicystic kidneys | |||
| Hydronephrosis | |||
| Joint flexion deformities | |||
| Mental retardation |
In some instances, skeletal dysplasias with associated thoracic anomalies can result in secondary pulmonary hypoplasia. In addition to severe and universally fatal skeletal dysplasias, such as achondrogenesis, osteogenesis imperfecta type 2, a few short-rib polydactyly syndromes, and thanatophoric dysplasia, there are many less severe skeletal dysplasias that may result in pulmonary compromise and respiratory difficulties at or shortly after birth. The long-term prognosis for each depends on the extent of the pulmonary hypoplasia and ability for the lungs to grow within the confines of the thoracic cage over time. Table 14-4 lists some of the skeletal dysplasias associated with respiratory difficulties at birth, but with the possibility of long-term survival.
| SYNDROME | CLINICAL FEATURES | MODE OF INHERITANCE | GENE OR LOCUS IF KNOWN |
|---|---|---|---|
| Camptomelic dysplasia | Pulmonary hypoplasia | Autosomal recessive versus autosomal dominant | Gene = SOX |
| Cystic hygroma, lymphedema | |||
| Acromelia with short bowed limbs, clubfoot, splayed toes | |||
| Polycystic kidneys | |||
| Asphyxiating thoracic dystrophy (Jeune syndrome) (incidence 1:100,000-130,000 live-born infants) | Pulmonary hypoplasia or insufficiency | Autosomal recessive | Unknown |
| Long, narrow thorax with rib and clavicular anomalies | |||
| Polydactyly | |||
| Hepatic, renal, and pancreatic cysts | |||
| Autosomal recessive spondylocostal dysostosis (Jarcho-Levin syndrome) | Pulmonary insufficiency | Autosomal dominant | Gene = DLL3 at 19q13; MESP2 ; LFNG |
| Vertebral and rib anomalies with a crablike chest | |||
| Short neck with normal limbs | |||
| Spondylocostal dysostosis with anal atresia and urogenital anomalies | Pulmonary hypoplasia | Autosomal recessive | Unknown |
| X-ray findings similar to Jarcho-Levin syndrome | |||
| Single umbilical artery | |||
| Internal and external genitourinary anomalies | |||
| Hydronephrosis | |||
| Spondyloepiphyseal dysplasia congenita | Pulmonary insufficiency secondary to restrictive lung disease | Autosomal dominant | Gene = COL2A1 |
| Barrel chest with platyspondylisis | |||
| Scoliosis, kyphosis, lordosis | |||
| Pectus carinatum | |||
| Coxa vara and clubfeet | |||
| Myopia and retinal detachment | |||
| Cleft palate | |||
| Hypotonia | |||
| Rhizomelic chondrodysplasia punctata type 1 | Pulmonary insufficiency | Autosomal recessive | Gene = PEX7 |
| Rhizomelic limb shortening with epiphyseal calcifications | |||
| Coronal vertebral clefts and kyphoscoliosis | |||
| Microcephaly with seizures, mental retardation, or cortical atrophy | |||
| Sensorineural hearing loss | |||
| Congenital cataracts | |||
| Often lethal before age 2 years |
Severe neuromuscular disorders associated with lack of fetal movement (fetal akinesia) may cause a host of problems in addition to pulmonary hypoplasia. Prenatal history is often significant for polyhydramnios (believed to be secondary to decreased fetal swallowing), joint contractures, and clubfeet. Whether the fetal akinesia is secondary to an inherent muscle or peripheral nerve problem, or instead a central nervous system problem, the clinical presentations are similar. Severe, congenital forms of inborn errors of metabolism, myopathies, or neuropathies may be clinically indistinguishable, unless specific diagnostic tests are done. Table 14-5 summarizes genetic conditions with neuromuscular underpinnings that may result in secondary pulmonary hypoplasia.
| SYNDROME | CLINICAL FEATURES | MODE OF INHERITANCE | GENE OR LOCUS IF KNOWN |
|---|---|---|---|
| Pena-Shokeir syndrome (fetal akinesia deformation sequence) | Pulmonary hypoplasia | Autosomal recessive (about 50% of cases). Empirical recurrence risks for siblings may be close to 10–15% | Unknown—may represent many different genetic syndromes causing suboptimal fetal movement |
| Generalized joint contractures with clubfeet | |||
| Polyhydramnios | |||
| Intrauterine growth restriction | |||
| Facial dysmorphism | |||
| Small thorax and thin ribs | |||
| Hydrocephaly with or without cerebellar hypoplasia | |||
| 30% are stillborn | |||
| Frequently lethal in neonatal period | |||
| Multiple pterygium syndrome (Escobar variant) | Pulmonary hypoplasia | Autosomal recessive | Gene = CHRNG |
| Eventration of diaphragm | |||
| Reduced facial movements | |||
| Severe joint contractures with pterygia (webbing) of joints and neck | |||
| Pathognomonic indentations over elbows and knees | |||
| Reduced muscle mass | |||
| Genitourinary anomalies | |||
| Stuve-Wiedemann syndrome | Pulmonary hypoplasia | Autosomal recessive | Gene = LIFR |
| Pulmonary hypertension | |||
| Short neck; short stature | |||
| Dysphagia | |||
| Thin ribs, osteoporosis | |||
| Progressive scoliosis | |||
| Short, thick long bones | |||
| Flexion contractures of fingers, elbows, and knees | |||
| Clubfeet | |||
| Hypotonia | |||
| Normal intelligence | |||
| Marden-Walker syndrome | Pulmonary hypoplasia | Autosomal recessive | Unknown |
| Prenatal and postnatal growth restriction | |||
| Microcephaly with CNS malformations | |||
| Hypotonia and decreased muscle mass | |||
| Fixed facial expression with dysmorphism | |||
| Joint contractures, clubfeet | |||
| Radioulnar synostosis | |||
| Scoliosis, kyphosis | |||
| Renal microcysts and dysplasia | |||
| Cryptorchidism | |||
| Spinal muscular atrophy type 1 | Pulmonary hypoplasia (in about ¼ of infants with spinal muscular atrophy type 1) | Autosomal recessive. Carried by about 1:40 otherwise healthy individuals | Gene = SMN1 |
| Above due to prenatal onset of hypotonia | |||
| Joint contractures | |||
| Feeding difficulties | |||
| Perinatal lethal Gaucher disease | Pulmonary hypoplasia | Autosomal recessive | Gene = GBA (enzyme acid β-glucosidase) |
| Severe fetal hydrops (30% stillborn) | |||
| Polyhydramnios | |||
| Fetal akinesia | |||
| Hypertonia; joint contractures | |||
| Microcephaly | |||
| Facial dysmorphism | |||
| Small thorax | |||
| Cardiomegaly | |||
| Collodion skin | |||
| Usually lethal by age 3 mo |
Congenital Diaphragmatic Hernia
CDH is a common anomaly and frequently associated with secondary pulmonary hypoplasia. CHD is seen in approximately 1 in 2000 live-born infants. This birth defect occurs as an isolated event and in association with more than 15 genetic syndromes ( Table 14-6 ). CDH also is seen with numerous cytogenetic deletion and duplication syndromes as one of many congenital anomalies; 15% of infants with CDH have a structural or numeric cytogenetic abnormality. Prenatal or postnatal detection of a CDH would require a thorough evaluation to look for dysmorphic features or additional birth defects that may be diagnostic of a specific syndrome.
| SYNDROME | CLINICAL FEATURES | MODE OF INHERITANCE | GENE OR LOCUS IF KNOWN |
|---|---|---|---|
| Congenital diaphragmatic hernia 1 | Diaphragmatic hernia alone | Autosomal dominant | Gene = ? NR2FZ ; locus = 15q26.1-q26.2 |
| Congenital diaphragmatic hernia 2 | Diaphragmatic hernia alone | Autosomal recessive | Locus = 8p23.1 |
| Congenital diaphragmatic hernia 3 | Diaphragmatic hernia alone | Autosomal dominant | Gene = ZFPM2 (8q22.3) |
| Anterior diaphragmatic hernia | Anterior diaphragmatic hernia | X-linked or multifactorial M > F | Unknown |
| Cornelia de Lange syndrome (Brachmann-de Lange syndrome) | Diaphragmatic hernia | Autosomal dominant | Gene = NIPBL |
| Prenatal growth restriction | |||
| Abnormal cry at birth | |||
| Microcephaly and classic facial dysmorphism | |||
| Congenital heart defects | |||
| Oligodactyly and other limb defects | |||
| Mental retardation | |||
| Fryns syndrome | Diaphragmatic hernia | Autosomal recessive. Two recognized microdeletions | Microdeletions = 15q26.2 and 8p23.1 |
| Large for gestational age | |||
| Coarse facies with excess hair | |||
| Small thorax | |||
| Distal limb and nail hypoplasia | |||
| Gastrointestinal, genitourinary, and CNS malformations | |||
| Often stillborn or die as neonates | |||
| Meacham syndrome | Diaphragmatic hernia | Unknown versus de novo autosomal dominant | Unknown |
| Pulmonary hypoplasia | |||
| Pulmonary rhabdomyomatous dysplasia | |||
| Congenital heart defects | |||
| Males with ambiguous genitalia or sex reversal | |||
| Simpson-Golabi-Behmel syndrome | Diaphragmatic hernia | X-linked recessive versus X-linked semidominant | Gene = GPC3 |
| Lung segmentation defects | |||
| Prenatal and postnatal overgrowth | |||
| Bulldog facies; macroglossia | |||
| Accessory nipples; 13 rib pairs | |||
| Cardiac and gastrointestinal defects | |||
| Large, cystic kidneys | |||
| Umbilical hernia | |||
| Polydactyly, syndactyly | |||
| Clubfoot | |||
| Embryonal tumors | |||
| Focal dermal hypoplasia (Goltz syndrome) | Diaphragmatic hernia | X-linked dominant (lethal in males) | Unknown |
| Ocular, dental, gastrointestinal, and genitourinary defects | |||
| Syndactyly, polydactyly | |||
| Cutaneous linear or patchy atrophy; papillomas with or without telangiectasias | |||
| Absent or hypoplastic nails | |||
| Autosomal recessive cutis laxa | Diaphragmatic hernia | Autosomal recessive | Unknown |
| Prenatal and postnatal growth restriction | |||
| Cutis laxa | |||
| Emphysema and cor pulmonale | |||
| Gastrointestinal and genitourinary diverticula | |||
| Joint laxity | |||
| Arterial tortuosity | |||
| Aneurysms | |||
| Hernias | |||
| Decreased elastin fibers in dermis | |||
| Epidermolysis bullosa with diaphragmatic hernia | Diaphragmatic hernia | Autosomal recessive | Unknown |
| Epidermolysis bullosa | |||
| Reported infants died shortly after birth | |||
| Syndromic microphthalmia (MIDAS syndrome [microphthalmia, dermal aplasia, sclerocornea]) | Diaphragmatic hernia | X-linked dominant (lethal in males). Also X chromosome microdeletion | Gene = HCCS (Xp22.3) |
| Unilateral or bilateral microphthalmia | |||
| Linear skin defects on face and neck | |||
| Cardiac defects | |||
| Genital with or without anal anomalies | |||
| CNS malformations | |||
| Syndromic anophthalmia with mild facial dysmorphism and normal intrauterine growth | Diaphragmatic hernia | Autosomal recessive | Gene = STRA6 (15q24.1) |
| Pulmonary dysplasia even in absence of diaphragmatic defect | |||
| Bilateral anophthalmia | |||
| Cardiac and genitourinary anomalies | |||
| Blepharophimosis with unusual eyebrows (trichoglyphic) | |||
| Large, low-set ears | |||
| Thoracoabdominal syndrome (X-linked midline defects including pentalogy of Cantrell) | Diaphragmatic hernia | X-linked recessive or X-linked dominant | Locus = Xq25-q26 |
| Cleft lip with or without cleft palate | |||
| Cardiac defects | |||
| Omphalocele; ventral hernias | |||
| Hydrocephaly, anencephaly | |||
| Renal agenesis, hypospadias | |||
| Donnai-Barrow syndrome (diaphragmatic hernia, exomphalos, absent corpus callosum, hypertelorism, myopia, and sensorineural deafness) | Diaphragmatic hernia | Autosomal recessive | Unknown |
| Abnormal ears | |||
| Sensorineural deafness | |||
| Myopia, ocular defects | |||
| Cardiac defects | |||
| Ventral abdominal wall defects | |||
| Agenesis of the corpus callosum | |||
| Omphalocele, diaphragmatic hernia, and radial ray defects | Diaphragmatic hernia | Probable autosomal recessive | Unknown |
| Omphalocele | |||
| Single umbilical artery | |||
| Radioulnar synostosis with thumb anomalies | |||
| Facial dysmorphism | |||
| Abnormal ears | |||
| Scoliosis | |||
| Diaphragmatic defects, limb deficiencies, and ossification defects of the skull | Diaphragmatic hernia | Autosomal recessive versus autosomal dominant with gonadal mosaicism | Unknown |
| Omphalocele | |||
| Limb deficiencies with syndactyly | |||
| Cranial ossification defects | |||
| Denys-Drash syndrome (Wilms tumor and pseudohermaphroditism or true hermaphroditism) | Diaphragmatic hernia | Autosomal dominant (overlaps with WAGR and Frasier syndrome) | Gene = WT1 (11p13) |
| Ambiguous genitalia (M or F) | |||
| Gonadal dysgenesis (M or F) | |||
| Gonadoblastoma | |||
| Nephropathy, nephritic syndrome leading to renal failure | |||
| Wilms tumor | |||
| Agonadism with multiple internal malformations (PAGOD syndrome [pulmonary hypoplasia, hypoplasia of the pulmonary artery, agonadism, omphalocele, diaphragmatic defects and dextrocardia]) | Diaphragmatic hernia | Autosomal recessive | Unknown |
| Omphalocele | |||
| Female external genitalia without internal gonads | |||
| Dextrocardia | |||
| Cardiac defects | |||
| Hypoplasia of lung even without diaphragmatic defect | |||
| Perlman syndrome (renal hamartomas, nephroblastomatosis, and fetal gigantism) | Diaphragmatic hernia | Autosomal recessive | Unknown |
| Prenatal overgrowth | |||
| Polyhydramnios | |||
| Facial dysmorphism | |||
| Visceromegaly | |||
| Hyperinsulinism | |||
| Renal hamartomas | |||
| Nephroblastomatosis | |||
| Wilms tumor | |||
| Pallister-Killian syndrome (mosaic tetrasomy 12p) | Diaphragmatic hernia | Cytogenetic | Mosaic tetrasomy 12p in skin fibroblasts (often not seen in lymphocytes) |
| Coarse facies with a prominent forehead and micrognathia | |||
| Sparse eyebrows, large ears | |||
| Short, webbed neck | |||
| Cardiac and genitourinary defects | |||
| Ventral wall defects | |||
| Polydactyly | |||
| Profound mental retardation and seizures | |||
| Stillbirth and neonatal mortality common | |||
| Emanuel syndrome (supernumerary derivative 22 chromosome) | Diaphragmatic hernia | Cytogenetic (results from 3:1 segregation from a parent who is a balanced carrier of an 11q23-22q11 translocation) | Supernumerary chromosome—a derivative 22 with part of 11q attached |
| Prenatal growth restriction | |||
| Microcephaly | |||
| Preauricular tags and sinuses | |||
| Cleft palate | |||
| Cardiac, genitourinary, and anal defects | |||
| Mental retardation | |||
| Deletion 1q32.3-q42.3 | Diaphragmatic hernia | Cytogenetic | Deletion = 1q32.3-1q42.3 |
| Deletion 8p23.1 | Fryns phenotype | Cytogenetic | Microdeletion = 8p23.1 |
| Congenital diaphragmatic hernia 2 | |||
| Deletion 15q26.1 | Fryns phenotype | Cytogenetic | Microdeletion = 15q26.2 |
| Diaphragmatic hernia |
CDH arises early in gestation when the pleuroperitoneal membranes fail to seal the pleuroperitoneal canal completely. Of CDHs, 80% are left-sided and result in abdominal viscera sliding up into the pleural cavity ( Fig. 14-1 ). Postnatal survival reportedly ranges from 39% to 95% after repair of CDH. This large variation in mortality depends largely on the severity of the resulting pulmonary hypoplasia and abnormal pulmonary vasculature. If extracorporeal membrane oxygenation is required, the prognosis is not favorable. The overall outcome of CDH is recognized to be worse when it is found in association with certain syndromes, such as Fryns syndrome. A recurrence risk for sibs of a child with an isolated CDH, based on empiric data, is approximately 2%.

Isolated CDH is generally believed to be a sporadic occurrence, although autosomal dominant and autosomal recessive pedigrees have been described. Several loci and putative predisposition genes have been found in cases of CDH, with cytogenetic deletions, duplications, and translocations often uncovering a CDH locus (see Table 14-6 ).
Segmentation Defects and Heterotaxy
During the embryonic stage, the right and left bronchial buds begin to grow, subsequently dividing into secondary bronchi. Normally, the right lung bud divides into three segments, and the left lung bud divides into two segments. Rarely, segmentation or lobulation defects occur and may or may not be associated with other visceral heterotaxies. Nearly 80% of children with right isomerism (bilateral right lung) have asplenia, leading to a risk for overwhelming pneumococcal sepsis. A similar proportion with left isomerism (bilateral left lung) has multiple small spleens (polysplenia). A few genetic syndromes have been associated with these types of defects ( Table 14-7 ).
| SYNDROME | CLINICAL FEATURES | MODE OF INHERITANCE | GENE OR LOCUS IF KNOWN |
|---|---|---|---|
| Smith-Lemli-Opitz syndrome (RSH syndrome) (Incidence 1:20,000-40,000) | Incomplete lobulation of lung with or without lung hypoplasia | Autosomal recessive | Gene = DHCR7 (enzyme is 7-dehydrocholesterol reductase) |
| Prenatal growth restriction | |||
| Failure to thrive | |||
| Microcephaly, epicanthal folds | |||
| Broad alveolar ridges | |||
| Cardiac defects | |||
| Hypospadias and other genitourinary defects | |||
| Two to three toe syndactyly and polydactyly | |||
| Foot anomalies | |||
| Mental retardation, seizures | |||
| CNS defects | |||
| Pallister-Hall syndrome (hypothalamic hamartoblastoma, hypopituitarism, imperforate anus, and postaxial polydactyly) | Abnormal lung lobulation | Autosomal dominant. Most cases are sporadic | Gene = GLI3 (7p13) |
| Intrauterine growth restriction | |||
| Microphthalmia, abnormal ears | |||
| Buccal frenula, cleft lip/palate | |||
| Cardiac, genitourinary, and anal defects | |||
| Polydactyly, syndactyly | |||
| CNS hypothalamic hamartoblastoma with pituitary hypoplasia | |||
| Simpson-Golabi-Behmel syndrome | See Table 14-5 | See Table 14-5 | See Table 14-5 |
| Asplenia with cardiovascular anomalies (Ivemark syndrome) | Right isomerism (bilateral right lung) | Autosomal recessive | Locus = 11q13 |
| Asplenia | |||
| Severe cardiac defects (TAPVR) | |||
| Midline liver | |||
| Malrotation of the gut | |||
| Situs inversus viscerum | Left isomerism (bilateral left lung) | Autosomal recessive | Locus = 7p21 |
| Polysplenia | |||
| Intrauterine growth restriction | |||
| Cardiac defects (PAPVR) | |||
| Severe Rubinstein-Taybi syndrome (chromosome 16p13.3 deletion) | Abnormal lung lobulation | Cytogenetic. Contiguous gene syndrome, as opposed to a point mutation or nondeletion Rubinstein-Taybi syndrome | Microdeletion = 16p13.3; deletion size 400 kb to 3 Mb |
| Accessory spleens | |||
| Cardiac defects, especially hypoplastic left heart | |||
| Renal agenesis | |||
| Neonatal seizures |
Cystic Adenomatoid Malformations
Cystic adenomatoid malformation of the lungs is a developmental abnormality that results from overgrowth of the terminal respiratory bronchioles modified by intercommunicating cysts. Cystic adenomatoid malformation encompasses variably sized cysts that, as they enlarge, compress adjacent lung tissue ( Fig. 14-2 ). The reported incidence is 1 in 25,000 to 35,000. It is useful to divide these cystic lesions into large cyst and small cyst types. The changes that have long been associated with the “adenomatoid” type now are recognized in various conditions with large airway obstruction and are more accurately called “pulmonary hyperplasia.”


Large Cyst Type (Stocker Type 1)
The type 1, or large cyst lesion usually manifests in early infancy with progressive respiratory distress as the cystic region expandsby air trapping with compression of adjacent lung tissue and ultimately mediastinal shift. Occasionally, these lesions are so large that growth of the normal lung tissue is impaired, resulting with pulmonary hypoplasia related to this space-occupying lesion. It is the most common type of cystic adenomatoid malformation and has the best prognosis because these malformations are usually localized and affect only part of one lobe. Cysts are usually larger than 2 cm in diameter. They are lined by respiratory epithelium and have a wall resembling bronchioles with small amounts of smooth muscle, but no glands. Some of these lesions may contain mucigenic epithelium. Increased risks for neoplasias, most commonly bronchioloalveolar carcinoma, have been a rationale for the early surgical resection of these lesions even when there are no clinical symptoms. This relationship is presumed to be related to neoplastic change in the mucigenic epithelium, which frequently is found as a minor component of the large cyst type of cystic adenomatoid malformation.
Small Cyst Type (Stocker Type 2)
The small cyst lesion has been recognized widely in areas of lung in which there is airway obstruction during development, such as bronchial atresia, isolated and with systemic arterial connection and extralobar sequestration. On pathologic examination, these lesions show typical and distinctive features with regional replacement of the lung parenchyma by microcystic maldevelopment. Mucigenic epithelium is rare. Depending on the size and degree of pulmonary compromise, infants may or may not be symptomatic at birth. A small percentage may be asymptomatic until later in childhood and often manifest with recurrent pneumonia or chest pain. Cystic adenomatoid malformations are usually isolated occurrences and are not thought to be genetically determined.
Bronchogenic Cysts and Other Cystic Lesions
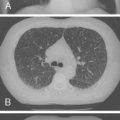
Foregut cysts are closed epithelium-lined sacs developing abnormally in the thorax from the respiratory tract and primitive developing gut. When these structures differentiate toward airway and contain hyaline cartilage plates in the wall, they are called “bronchogenic cysts,” whereas structures developing toward the gut are termed “enterogenous cysts.” Bronchogenic cysts are the most common cysts in infancy, although many do not manifest until adolescence or adulthood. Most are situated in the middle mediastinum close to the carina, but do not communicate with the trachea or bronchi. Less frequently, they are adjacent to the esophagus. They are usually single, unilocular cystic structures that are filled with fluid or mucus, and more commonly are located on the right ( Fig. 14-3 ). Because of inadequate drainage, such cysts may be associated with recurrent infections and may appear as a consolidation or area of atelectasis on chest x-ray. Bronchogenic cysts also may be found within the lung hilum and parenchyma. When located within the lung, they are identical in their gross and histologic appearance to cysts in the mediastinum.



Similarly located cysts with enteric-type mucosa, usually gastric or esophageal and without cartilage plates within their wall, are enteric duplication cysts. Esophageal cysts are more common. They are intramural and do not involve the mucosa. Gastroenteric cysts characteristically are not connected to the esophagus. In infants, they are a common cause of a posterior mediastinal mass. Neurenteric cysts are posterior mediastinal lesions, also lined by enteric-type mucosa, and with a pedicle that extends into the spinal canal. They are virtually always associated with vertebral defects in the region of this communication. Rarely, congenital cystic lesions may be seen in conjunction with genetic syndromes. There is a specific autosomal recessive disorder described with multiple peripheral lung cysts or fibrocystic pulmonary dysplasia as the primary feature ( Table 14-8 ).
| SYNDROME | CLINICAL FEATURES | MODE OF INHERITANCE | GENE OR LOCUS IF KNOWN |
|---|---|---|---|
| Cystic disease of the lung | Cystic pulmonary lesions | Autosomal recessive (especially common in Yemenite and other non-Ashkenazi Jews) | Unknown |
| Recurrent lung infections | |||
| Spontaneous pneumothorax in the neonate | |||
| Proteus syndrome (encephalocraniocutaneous lipomatosis) | Cystic pulmonary lesions | Autosomal dominant (some cases may be due to somatic mosaicism for a gene mutation) | Gene = PTEN (only some cases) |
| Macrocephaly | |||
| Hemihypertrophy with localized overgrowth | |||
| Variable cutaneous lesions, including lymphangiomas, lipomas, epidermal nevi, hemangiomas | |||
| High risk of thrombosis | |||
| Kyphoscoliosis | |||
| CNS malformations | |||
| Down syndrome (trisomy 21) | Characteristic subpleural cysts | Cytogenetic | Extra copy of chromosome 21 |
| Facial dysmorphism | |||
| Short, broad neck | |||
| Cardiac, gastrointestinal, and genitourinary defects | |||
| Brachydactyly, clinodactyly | |||
| Single palmar crease |
Related posts:
 Chronic Lung Disease of Infancy
Chronic Lung Disease of Infancy
 Pulmonary Manifestations of Human Immunodeficiency Virus (HIV) Infection
Pulmonary Manifestations of Human Immunodeficiency Virus (HIV) Infection
 Pulmonary Manifestations of Endocrine and Metabolic Diseases
Pulmonary Manifestations of Endocrine and Metabolic Diseases
 Pulmonary Manifestations of Rheumatoid Diseases
Pulmonary Manifestations of Rheumatoid Diseases
 Pulmonary Manifestations of Gastrointestinal Diseases
Pulmonary Manifestations of Gastrointestinal Diseases
 Pulmonary Manifestations of Dermatologic Diseases
Pulmonary Manifestations of Dermatologic Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


