Pulmonary complications are a common side effect of pediatric hematologic and oncologic disorders and their treatment. Respiratory signs and symptoms can be the presenting manifestations of several types of pediatric malignancies. The anatomic structures typically affected by this group of disorders are the mediastinum, airways (trachea and bronchus), alveolocapillary units, lung parenchyma, pleura, diaphragm, and chest wall. Many of these thoracic structures, as exemplified by the thymus, continue to develop intensively after birth and are common targets of malignant transformation. This chapter describes the pulmonary symptoms and management of pediatric hematologic and oncologic disorders that directly or indirectly affect the respiratory system.
Oncologic Diseases
Complications Related to Disease and Acute Treatment
Pediatric acute leukemias and lymphomas are clonal disorders of the blood-forming cells (erythroid, myeloid, lymphoid, and megakaryocytic) that result from the transformation of immature progenitor cells of these hematopoietic lineages. These disorders are grouped in four broad categories: acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), non-Hodgkin lymphoma (NHL), and Hodgkin disease (HD). They are classified further by the type of specific progenitor cells involved in the process. Because the hematopoietic system is functionally diverse and has a wide anatomic distribution, the clinical and biologic characteristics of leukemias and lymphomas in young patients vary substantially.
In the United States, of the approximately 13,000 new cases of cancer diagnosed per year in children, adolescents, and adults younger than 20 years old, 18.7% are ALL, 8.8% are HD, 6.5% are NHL, and 3.5% are AML. Of the neoplasias in children and adolescents, 50% are hematologic malignancies. The incidence of leukemias and lymphomas varies with age. ALL represents 17% of all cases of pediatric cancer in neonates and infants, 46% of cases of cancer in children 2 to 3 years old, and 9% of cases of cancer in individuals 15 to 20 years old. Although the incidence of AML also varies with age, the rates are highest in the first 2 years of life, after which they decrease to a nadir at the end of the first decade, and then gradually increase during the second decade of life. Conversely, the rates of NHL and HD increase with age. The incidence of HD increases with age, with 43.2 cases per 1 million for individuals 19 years old.
The pattern of age distribution among leukemia and lymphomas suggests that etiologic factors differ in these diseases. Generally, the process of malignant transformation of a specific hematopoietic progenitor cell is believed to occur during fetal development in acute leukemias and postnatally in lymphomas —hence, the incidence of lymphomas is greatly affected by environmental factors. In some areas of Africa, Burkitt lymphoma and Kaposi sarcoma are two common types of pediatric malignancy.
The intrathoracic cavity harbors primary (thymus) and secondary (lymph nodes and mucosa-associated lymphatic aggregates) lymphoid organs; thoracic lymphoid structures are commonly affected in lymphoid leukemias and lymphomas. Enlargement of the thymus or its associated lymph nodes is very common in T cell ALL, lymphoblastic NHL, and HD. Mediastinal involvement is extremely rare in AML. Chest radiography is an effective screening tool to identify children with mediastinal mass and should be obtained in any patient newly diagnosed with leukemia or lymphoma. A large mediastinal mass in a child should be considered a medical emergency and managed in a tertiary care hospital by a multidisciplinary team. Depending on the size, location, organ compression, and progression time, a particular constellation of clinical signs and symptoms indicates whether the airways, great vessels, or both are predominantly affected. A detailed assessment of the patient’s medical history and results of the physical examination can provide clues regarding whether the compression is affecting the airways, great vessels, heart, or more than one of these structures.
Tracheal Compression and Superior Vena Cava Syndrome
Large tumors in the anterior mediastinum can cause tracheal compression, superior vena cava (SVC) syndrome, or both. The most common tumors in this area are HD and NHL. Tracheal compression may cause cough, stridor, dyspnea, or orthopnea. In these cases, chest radiograph may reveal an anterior mediastinal mass, prominent hilar lymph nodes, posterior tracheal deviation, atelectasis, and pleural effusion. Tracheal compression is not always obvious, however, from the physical examination or chest radiograph. It may become apparent when a patient is unable to lie supine because of increased dyspnea; the patient should be considered at high risk for complete tracheal obstruction. The SVC is particularly susceptible to compression because it is surrounded by lymph nodes and is in direct contact with rigid anatomic structures. It has a delicate vessel wall and low intraluminal pressure.
Symptoms resulting from moderate to severe SVC compression include headache, dizziness, syncope, and cardiovascular collapse secondary to decreased venous return ( Fig. 7-1 ). Physical examination may be unremarkable or may show facial and periorbital edema, cyanosis, plethora, neck and chest vein distention, papilledema, and edema of the upper extremities. The severity of the clinical manifestations of SVC syndrome depends on how rapidly the obstruction arises, and whether sufficient time has elapsed for new collateral blood vessels to develop.


Children with tracheal compression or SVC syndrome are often anxious and diaphoretic. They resist efforts to be placed in the supine position and should not be forced into this position. Assessment of airway patency with computed tomography (CT) scan or flexible bronchoscopy is ill-advised because patients do not tolerate these procedures well and may be very difficult to resuscitate if they experience cardiorespiratory collapse. Total airway occlusion has been reported during the induction of general anesthesia, tracheal intubation or extubation, movement of a patient to a supine or flexed position (for lumbar puncture), and conscious sedation. Flow-volume loops have been used to indicate the degree of central airway obstruction and may be helpful in distinguishing fixed from variable intrathoracic airway lesions.
Whole-body positron emission tomography (PET) has been increasingly used to determine the extent of disease and response to therapy. NHL and HD are metabolically active, and so they can be detected with this method. Although PET/CT ( Fig. 7-2 ) is still considered a research tool in disease staging and prognosis, some studies have shown the superiority of PET/CT to PET or CT alone in the management of NHL and HD. The least invasive technique is used to obtain tissue for diagnosis. The diagnosis often can be made from bone marrow aspirate or peripheral lymph node biopsy, which can be obtained with topical anesthesia. Similarly, if pleural effusion is present, thoracentesis can be used in making a diagnosis ( Fig. 7-3 ).



A proper regimen of chemotherapy should be started as soon as a diagnosis is established. Intravenous access should be started in the lower extremities because most of the venous return proceeds through the inferior vena cava. Although SVC syndrome per se does not represent a medical emergency, most children with SVC syndrome experience some degree of airway compression. Therapy with radiation to the mid-plane or corticosteroids (hydrocortisone, 2 mg/kg every 6 hours) or both usually alleviates symptoms. Most mediastinal masses in children and adolescents are highly sensitive to chemotherapy. These patients are at high risk of uric acid nephropathy and consequently kidney dysfunction. Hyperhydration and alkalinization used for patients at high risk of tumor lysis syndrome (hyperuricemia) should be avoided in patients with SVC syndrome, however, because of the risk of respiratory insufficiency and worsening of existent pleural effusion. Rasburicase is very effective in rapidly decreasing plasma urate, avoiding the need for hyperhydration and alkalinization.
Doppler ultrasonography and echocardiography may be indicated for evaluating flow through the great vessels and myocardial function. If thrombosis is detected, use of unfractionated or low-molecular-weight heparin should be considered to prevent propagation of the thrombus. In rare cases, catheter-directed thrombolysis has been performed to relieve obstruction. Most importantly, prompt specific chemotherapy for primary malignancy should be instituted as soon as the diagnosis is established.
Pulmonary Leukostasis Syndrome
Approximately 50% of children with ALL and AML present with an abnormally elevated blood leukocyte count. Severe leukocytosis (defined as peripheral leukocytes ≥100,000/mm 3 ) occurs in approximately 20% of children with ALL and 15% of children with AML. Most of these patients have no pulmonary signs or symptoms associated with this abnormality. Depending on the number of leukocytes and the type of leukemia, however, a constellation of signs and symptoms, known as pulmonary leukostasis syndrome, may occur. Leukostasis can affect any organ, but it most commonly involves brain and lungs. Clinically, pulmonary involvement is characterized by shortness of breath, tachypnea, dyspnea on exertion, and pleuritic chest pain. Mortality in patients with leukostasis is approximately 20%, usually secondary to pulmonary or intracranial hemorrhage.
Patients with AML tend to develop symptomatic leukostasis at much lower leukocyte counts than do patients with ALL. Postmortem evidence of leukostasis in AML, including extensive leukemic infiltration of the alveoli and parenchyma, and occlusion of small pulmonary vessels, has been found in patients presenting with a wide range of leukocyte counts. Symptomatic pulmonary leukostasis occurs almost exclusively in children with AML.
Respiratory distress and early death resulting from hyperleukocytosis (defined as leukocyte counts ≥200,000/mm 3 ) is more common in children with AML, occurring in 6% of patients. A high initial white blood cell count seems to be the only marker associated with the development of respiratory complications. In addition, because myeloblasts express adhesion molecules that attach to the pulmonary vascular endothelium, they are more tissue-invasive than lymphoblasts. These factors apparently are more relevant to the severity of pulmonary leukostasis syndrome than the number of leukemia cells. The adverse interactions between leukemic cells and organs such as lungs, brain, and kidneys seem to be worsened further in M5 AML and M4 AML because these subtypes of AML release many proinflammatory cytokines and lysozymes.
Chest radiographs can be normal, or they may reveal variable degrees of diffuse alveolar-interstitial infiltrates and pulmonary vessel enlargement. Findings of chest CT scans include bilateral thickening of the interlobular septa with patchy areas of ground-glass opacity that resemble that of interstitial edema. A ventilation/perfusion lung scan can show mismatched defects, but is not specific for leukostasis, and reliance on it can lead to the misdiagnosis of pulmonary embolism. Imaging studies are nonspecific for pulmonary leukostasis.
Hypoxemia in patients with hyperleukocytosis can be a true or false finding. Causes of true hypoxemia in these patients include infection, hemorrhage, pulmonary embolism, pulmonary alveolar proteinosis (PAP), and occlusion of the pulmonary vasculature by leukocyte thrombi. False hypoxemia can result from technical inaccuracies in the measurement of PaO 2 , and often may reflect that leukemic cells are metabolically active in vitro . In this situation, low levels of PaO 2 reflect consumption of dissolved oxygen by leukocytes or platelets after the arterial blood specimen is obtained, a phenomenon called “oxygen steal.” Consistent with this concept is the observation that PaO 2 values are negatively correlated with the number of circulating leukocytes. Because pulse oximetry measures capillary oxygen instead of whole-blood oxygen saturation, this method is considered the most accurate way to assess oxygenation in patients with hyperleukocytosis.
Acute leukemia complicated by severe hyperleukocytosis should be considered a medical emergency. Admission of the patient to an intensive care unit and proper management of aggravating factors, such as dehydration, electrolyte or metabolic imbalances, infection, thrombocytopenia, and coagulopathy, should be initiated immediately. Fluid balance is required to avoid excessive fluid retention. Transfusion of platelet concentrate or fresh frozen plasma is used to reduce the risk of bleeding. Prophylaxis of thromboembolic phenomena with heparin is not indicated. Transfusion of packed red blood cells should be withheld or be given with caution to prevent an increase in blood viscosity before the reduction of the leukocyte count. Anemia in patients with hyperleukocytosis can be seen as an adaptive mechanism. Inhaled nitric oxide to produce pulmonary vasodilation and ventilation/perfusion mismatch reduction has been used by some investigators. Leukapheresis or exchange transfusion (for young children) is commonly considered as a temporary measure until specific antileukemia treatment is initiated. The efficacy of leukapheresis in reducing mortality or life-threatening complications has not been evaluated in controlled clinical trials.
Pulmonary Alveolar Proteinosis
PAP can rarely be a cause of respiratory failure in patients with acute leukemia, particularly AML. PAP is characterized by the intra-alveolar accumulation of surfactant proteinaceous material, resulting in impaired alveolocapillary gas exchange ( Fig. 7-4 ). Three types of PAP have been recognized: congenital, associated with or secondary to systemic disease, and idiopathic. The last-mentioned is the most common form of PAP and believed to have an autoimmune basis.

The mechanism of PAP has not been firmly established, although surfactant homeostasis apparently is impaired in this disease. Although only 10% of surfactant comprises proteins, denominated A, B, C, and D, they are crucial to surfactant metabolism, opsonization of microorganisms, and stimulation of alveolar macrophage. Alveolar type II epithelial cells and alveolar macrophage control surfactant synthesis, storage, and catabolism. Alveolar macrophages, which are produced by bone marrow hematopoietic cells, internalize and catabolize surfactant by a process believed to depend on granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling. Several lines of evidence suggest that abnormality of the GM-CSF signaling in the lung is associated with PAP pathogenesis. First, histologically similar PAP lung changes occur in mice genetically deficient in GM-CSF or its receptor. Abnormal clearance of surfactant lipids is impaired in GM-CSF–deficient mice, providing an explanation for the intra-alveolar accumulation of PAS-positive material. PAP in GM-CSF–deficient mice can be corrected by the local expression of GM-CSF in the lungs, or by bone marrow transplantation from a wild-type mouse. Second, PAP occurs in individuals harboring constitutional mutations in the genes encoding surfactant proteins B or C or the β c chain of the GM-CSF receptor. Finally, autoantibodies that bind and neutralize GM-CSF are found in bronchoalveolar lavage (BAL) of patients with the idiopathic form of PAP.
Imaging studies of the lungs suggest PAP. The chest radiograph usually shows bilateral airspace disease characterized as ground-glass opacities suggestive of pulmonary edema, but without other signs of left-sided heart dysfunction. There is no relationship between clinical and radiographic findings, with the latter being disproportionately more severe. High-resolution CT scans reveal a pattern referred to as “crazy-paving,” which is characterized by ground-glass opacifications with superimposed interlobular septal and intralobular thickening.
BAL specimens can be used to establish the diagnosis. Lung biopsy is the gold standard for the diagnosis of PAP, but it sometimes can yield false-negative results.
The incidence of PAP associated with leukemia is unknown, but a study by Cordonnier and colleagues suggested that it can be 10% in patients with leukemia and respiratory failure. Treatment of PAP consists of overall supportive care because many of these patients have severe neutropenia from previous chemotherapy, and whole-lung lavage for patients with severe respiratory distress. Most cases of chemotherapy-induced PAP usually improve with the resolution of neutropenia.
Because the mechanism of PAP has been associated with GM-CSF deficiency, administration of GM-CSF has been proposed as a treatment of PAP. In a study of 25 patients with idiopathic PAP, subcutaneous administration of GM-CSF improved oxygenation and decreased alveolar-arterial oxygen gradient in approximately 50% of the patients. Whether GM-CSF has a therapeutic value in children with leukemia and respiratory distress associated with clinical and laboratory features of PAP remains to be determined. Defective expression of GM-CSF/interleukin-3/interleukin-5 receptor common β chain has been shown to occur in children with AML associated with respiratory failure.
Solid Tumors
Solid malignancies in children differ markedly from such tumors in adults; biology and histology define a very distinct group of neoplasms, most of them of dysontogenetic origin, with unique clinical manifestations and natural history. Epithelial malignancies are extremely rare in children; sarcomas and dysontogenetic tumors constitute most solid cancers. Generally, these groups of malignancies are aggressive, and systemic dissemination (microscopic or macroscopic) is present at the time of diagnosis. For this reason, an extensive metastatic work-up is necessary, and most malignancies require systemic therapy regardless of the local stage.
Osteosarcoma
Osteosarcoma, a malignant neoplasm derived from primitive mesenchymal cells and characterized by the presence of osteoid-producing spindle cell stroma, is the most common malignant bone tumor in pediatric patients. Osteosarcoma accounts for 2.6% of all neoplasms in children, with an estimated annual incidence of 3.9 per 1 million in white children and 4.5 per 1 million in African-American children. Most osteosarcomas occur during the first 2 decades of life, a period characterized by rapid skeletal growth. Overt macroscopic metastatic disease is seen in a significant proportion of patients, and has a grave prognosis when present. The most common site for metastatic spread is the lung; 14% to 24% of patients have macroscopic pulmonary disease at the time of diagnosis. Approximately 5% of all osteosarcomas are multifocal (i.e., involving two or more bones at the time of diagnosis). Multifocal osteosarcoma has a more aggressive clinical behavior, a higher incidence of pulmonary metastases, and a very poor prognosis.
When osteosarcoma is diagnosed, even the most accurate staging procedures detect metastases in only a few patients. Without adequate treatment, however, most patients with seemingly localized disease develop secondary metastases within 1 year. This finding implies that micrometastases are already present at diagnosis in most patients.
With appropriate treatment, 60% to 70% of patients with localized disease are expected to be cured. The outcome of patients with clinically detectable metastatic disease at diagnosis is usually very poor; less than 30% of patients are expected to survive. Care of these patients needs to combine a multimodal approach, with intensive preoperative and postoperative chemotherapy and resection of the primary tumor and all the metastatic lesions. Using these guidelines, more recent research reports 2- to 5-year, progression-free survival rates of 25% to 45%. Factors that affect outcome include number of metastases, laterality, and the ability to perform a complete resection.
The lungs also are the most common sites of treatment failure in patients with osteosarcoma. For patients with recurrent disease, a surgical approach is recommended; the 5-year postrelapse survival of patients in whom a complete resection of all macroscopic disease can be achieved is almost 40%, whereas it is 0% for patients with unresectable disease. Prognostic factors for survival after recurrence include isolated lung metastases, late (>24 months) recurrences, and small number of pulmonary nodules.
Plain chest radiographs detect lung metastases in most cases. High-resolution CT of the chest is the procedure of choice, however. False-positive results occur, particularly with smaller lesions, and biopsy confirmation of the lung disease is usually required. Osteoid matrix produced by osteosarcoma cells forms bone and causes calcification in pulmonary nodules. Seventy-five percent of metastatic osteosarcoma nodules lack calcification, however. Conversely, only 50% of calcified nodules in newly diagnosed patients with osteosarcoma are malignant, whereas 65% of noncalcified lesions are found to be metastatic osteosarcoma. Mediastinal adenopathies are very rare in patients newly diagnosed with osteosarcoma, and their presence in the context of lung nodules favors the diagnosis of a nonmalignant condition; 60% of patients with benign nodules have mediastinal disease compared with less than 20% of patients with metastases.
CT is often unable to distinguish benign from malignant pulmonary disease, and a surgical procedure is often required for confirmation. CT can miss 40% to 50% of the lesions that are found later during the surgical procedure. This finding highlights the importance of manual palpation during open thoracotomy; minimal access procedures such as thoracoscopy should not be the approach if the goal is resection.
In patients who have undergone thoracotomy for metastatic disease, new nodules are likely to be metastatic, and surgical intervention is warranted. Surgical scarring often makes the diagnosis of recurrent disease difficult, however. McCarville and associates evaluated the imaging patterns of recurrent nodules after thoracotomy in patients with osteosarcoma. Pulmonary nodules recurred in 90% of patients who underwent thoracotomy, and metastatic disease was found in 90% of patients who underwent another surgical procedure. Only one third of the nodules appeared to be calcified on CT scans. A consistent pattern suggestive of malignancy was the presence of progressive pleural thickening. In contrast to newly diagnosed patients, in most patients who had undergone thoracotomy, recurrent pulmonary disease was associated with the presence of mediastinal adenopathies. With subsequent recurrence, the proportion of malignant lesions and the incidence of malignant mediastinal disease increased. Also, half of the new lesions occurred in previous scars, and the estimated probability that a recurrent nodule in the site of a scar was malignant was 82%.
The ability to control the pulmonary micrometastatic disease after completion of therapy would result in a significant improvement in outcome. Muramyl tripeptide phosphatidylethanolamine (MTP-PE), a synthetic lipophilic analogue of muramyl dipeptide (a component of the cell wall of bacille Calmette-Guérin), has been encapsulated in liposomes to deliver the agent selectively to monocytes and macrophages to activate their tumoricidal properties. In an animal model, the administration of liposome-encapsulated muramyl tripeptide (L-MTP-PE) resulted in activation of pulmonary macrophages and eradication of pulmonary micrometastases. In the cooperative Children’s Cancer Group and Pediatric Oncology Group intergroup INT0133 clinical trial, patients with localized osteosarcoma received standard treatment with methotrexate-cisplatin-doxorubicin regimen. Patients were first randomly assigned to receive ifosfamide, L-MTP-E, or a combination of both. Adding a combination therapy with ifosfamide and L-MTP-PE to the standard regimen resulted in a significantly better outcome than the other three regimens. This improved outcome could be due to the synergistic effect of L-MTP-PE and ifosfamide.
It also is possible to induce activation of alveolar macrophages by nebulization of GM-CSF. Based on encouraging preliminary data, the Children’s Oncology Group is currently evaluating this approach in patients with metastatic osteosarcoma. Another similar approach is the interleukin-12 gene transfer by aerosol using a nonviral vector, such as polyethylenimine, a polycationic DNA carrier. Interleukin-12 has a well-known activity against various tumors. The systemic administration of interleukin-12 is limited by its toxicity. In animal models, aerosol therapy resulted in a significant decrease in the size and number of metastatic nodules. Interleukin-12 also has been shown to enhance the sensitivity of osteosarcoma cells in vitro to 4-hydroxy-cyclophosphamide by a mechanism involving the Fas pathway, which suggests that this approach may act synergistically with ifosfamide. The same polyethylenimine transfer technology has been shown to provide an antitumor effect using p53 transfection.
Aerosolized technology also has been developed to deliver chemotherapeutic agents directly to the lungs. In an animal model, aerosolized 9-nitrocamptothecin liposome was administered to mice that had subcutaneous xenografts of various human cancers and to mice with lung metastases of osteosarcoma. The results were encouraging; a significant antitumor effect was noted in the xenografts and in the pulmonary disease, suggesting not only a local, but also a systemic effect. Clinical trials to assess this form of therapy are under way.
Ewing Sarcoma Family of Tumors
The term “Ewing sarcoma family of tumors” (ESFT) refers to a group of small round cell neoplasms of neuroectodermal origin. Ewing sarcoma of the bone is the least differentiated form, and primitive neuroectodermal tumor and peripheral neuroepithelioma are the most differentiated forms. Of all types of pediatric malignancy, 3% are ESFT; these tumors are rare in nonwhites. Approximately 40% of all bone cancers in children are ESFT of the bone, the second most common type of bone malignancy in children after osteosarcoma. Patients with ESFT commonly present with symptoms during the second decade of life; 80% of patients with EFST are younger than 18 years, and the median age at diagnosis is 14 years. ESFT has a tendency to involve the shaft of long tubular bones, the pelvis, and the ribs, but almost every bone can be affected. More than 50% of the tumors arise from axial bones, with the pelvis being the most commonly involved (23% to 27%). Primary ESFT of the chest wall, previously known as Askin tumor, occurs in 12% of patients with ESFT.
ESFT are aggressive neoplasms; systemic manifestations including fever and anemia are present in 10% to 15% of patients, and approximately 20% to 25% of cases have clinically apparent metastatic disease at the time of diagnosis. Metastatic disease seems to be associated with older age and large tumors or with pelvic primary tumors. Isolated lung disease, usually bilateral, occurs in 25% to 45% of patients with metastases; most (50% to 60%) have extrapulmonary disease (usually of the bone and bone marrow). Overall, approximately 10% of patients have lung metastases at the time of diagnosis ( Fig. 7-5 ).

The most important prognostic factor is the presence of metastatic disease at diagnosis. Advances in the treatment of ESFT have resulted in only a very modest improvement in the survival of patients with metastases. Even among patients with metastatic disease, however, there is some heterogeneity. Patients with isolated lung metastases may have a better prognosis than patients with extrapulmonary metastases, with long-term survival rates approaching 40% to 45%. Among patients with lung metastases, patients with unilateral disease and patients with good histologic response to induction chemotherapy seem to have a survival advantage.
In approximately 50% of patients with isolated lung metastases, treatment failures occur as isolated pulmonary disease again, suggesting that further response consolidation might improve the outcome of these patients. In contrast to osteosarcoma cells, ESFT cells are very sensitive to radiation therapy, and lung radiation is an alternative to lung surgery. In the first American Intergroup Ewing Sarcoma Study, lung radiation was associated with a lower incidence of lung (and distant) recurrences. With the development of more intensive chemotherapeutic regimens, lung radiation of localized disease was abandoned. It remains a therapeutic option, however, for patients with metastatic disease. In this regard, whole-lung radiation seems to provide a modest survival advantage in patients with metastatic disease. Preliminary data of the European Bone Marrow Transplant Registry suggest that an alternative approach to the treatment of patients with isolated lung metastases may be high-dose chemotherapy using a busulfan-based regimen and autologous stem cell rescue.
Patients with primary ESFT of the chest wall represent a distinct group. Many of these patients have infiltration of the pleura, have pleural effusion, or may develop intraoperative contamination of the pleural space. They may be at high risk of disease relapse within the pleural compartment, and many groups have used hemothorax radiation covering the entire pleural space. The use of hemithorax radiation seems to reduce systemic relapse rates (mainly in lung metastases) and to provide a survival advantage.
Soft Tissue Sarcomas
Pediatric soft tissue sarcomas (STS) are broadly divided into rhabdomyosarcoma (RMS) (40% to 45%) and non-RMS soft tissue sarcomas (NRSTS) (55% to 60%), which include several histologic subtypes, each one of which constitutes less than 5% to 10% of STS. Altogether, STS represent approximately 6% to 7% of the cases of malignancy in individuals younger than 20 years. The incidence of RMS is highest in infancy and during the first years of life, and it decreases and levels out at later ages, whereas NRSTS have their highest incidence in adolescence, although a peak is observed in infants.
RMS recapitulates the phenotypic and biologic features of developing skeletal muscle. Two broad categories are identified: Embryonal RMS with its botryoid and spindle cell variants accounts for two thirds of the cases, and alveolar RMS and undifferentiated RMS account for the remaining cases. RMS is clinically heterogeneous; it can arise anywhere in the body where mesenchymal tissue is found. One third of the cases arise in the head and neck region (orbit, parameningeal, and nonparameningeal sites); 25% arise in the genitourinary tracts, primarily bladder and prostate; and 18% arise in the extremities. RMS occur much less frequently in the trunk and pelvis.
Approximately 10% to 15% of patients with RMS have metastatic disease at the time of diagnosis. The incidence of metastatic disease is higher in patients with alveolar RMS (25% to 30%) than in patients with embryonal RMS (5% to 10%). Most cases of alveolar RMS are characterized by the presence of the t(2;13)(q35;q14) or, less frequently, t(1;13)(p36;q14) translocations, which result in the PAX3-FKHR or PAX7-FKHR chimeric genes. Patients with the PAX3 translocation have more widespread metastatic disease and a worse outcome. The PAX7 translocation seems to be associated with a lower incidence of lung metastases.
Approximately 5% of patients with RMS have lung metastases at the time of diagnosis. Only 15% to 25% of patients with metastatic disease have isolated lung metastases; in most cases, the presence of lung disease is a sign of more widespread metastatic RMS. The presence of isolated lung metastases is associated with favorable features, such as embryonal histology and negative nodal involvement. Patients with metastatic disease have a generally poor prognosis, with long-term survival estimates of less than 30%. Patients with isolated lung metastases seem to benefit from lung radiation, however, and have a slightly better outcome, with 4-year overall survival rates of 40%.
NRSTS comprise a very heterogeneous group of neoplasms of mesenchymal origin. The most common subtypes in children are dermatofibrosarcoma protuberans, synovial sarcoma, malignant fibrous histiocytoma, fibrosarcoma, and malignant peripheral nerve sheath tumor. Because each tumor is individually rare in pediatric patients, little is known about their biology and natural history in children. Most have clinical behavior similar to tumors in adults; however, there are notable exceptions, such as infantile fibrosarcoma and infantile hemangiopericytoma, neoplasms that are associated with characteristically good prognosis despite aggressive histologic features.
Lung metastases are present in 5% to 10% of the cases at diagnosis. Only 12% of children who have undergone gross tumor resection experience metastatic tumor recurrence. Patients with large (≥5 cm), invasive, or high-grade tumors have a significantly higher (25% to 35%) risk of distant disease recurrence, however, usually in the lungs. If patients are at high risk of distant metastases after local control or have unresectable or metastatic tumors at presentation, chemotherapy must be considered.
Among all the histologic subtypes of NRSTS, alveolar STS warrants special mention. This rare tumor accounts for only 1% of all STS. Of patients with this form of sarcoma, 65% have lung metastases at the time of diagnosis, however. This sarcoma has a very indolent course, and even in the presence of metastases, 5-year overall survival rates exceed 70% to 80%.
Wilms Tumor
Wilms tumor is the most common childhood renal tumor, accounting for approximately 6% of all pediatric malignant disease. The most common presentation is an asymptomatic abdominal mass in a young child (typically <5 years old), although approximately one third of such patients present with abdominal pain, anorexia, and malaise. Hypertension is present in 25% of the cases, and congenital anomalies, such as aniridia, genitourinary malformations, or hemihypertrophy, are present in 10% to 20% of children. Histologic characteristics are the most important prognostic indicators; anaplasia (which occurs in 10% of cases) is associated with an adverse outcome. Lung metastases are present in approximately 10% of patients with favorable histology and in 20% of patients with anaplasia. In contrast to most other types of pediatric malignancy, Wilms tumor with lung metastases has a good outcome, with 5-year overall survival estimates exceeding 80% for patients with favorable histology. Lung radiation is an integral component in the management of these patients with lung metastases.
Plain chest radiographs have been used to define the presence of metastatic disease in patients with Wilms tumor. In approximately 10% of patients with lung nodules, plain chest x-rays are normal, however, and the lesions are visible only on CT scan. Nonmalignant nodular lesions are frequently identified on chest CT scans, but not on plain radiographs in children. In adults, 50% to 60% of lesions identified on CT only are not malignant. For these reasons, the role of lung radiation in this group of patients with CT-only lung nodules is unclear. In such patients treated on the National Wilms Tumor Studies 3 and 4, the 4-year event-free survival estimates were 89% for patients receiving radiation and 80% for patients treated with chemotherapy only. Similarly, results of a study by the International Society of Pediatric Oncology showed that patients with lung nodules identified only on CT and who achieved a complete response to chemotherapy had a 5-year overall survival estimate of 83%. These results contrast with the findings of the United Kingdom Children’s Cancer Study Group, which indicated much lower (65%) survival rates if no radiation was used.
Because this issue remains unresolved, future trials by investigators in the Children’s Oncology Group are expected to assess the necessity of lung radiation for patients whose tumors have favorable biologic and histologic features and show rapid response to chemotherapy. It is possible that some children with stage IV favorable histology Wilms tumor may be treated successfully without lung radiation. Finally, an important consideration is the frequent extension of Wilms tumor into the renal vein and proximal inferior vena cava, which is sometimes associated with pulmonary embolism. In most cases, tumor thrombus can be removed en bloc with the kidney. If the tumor extends to the hepatic level or into the atrium, however, the risk of operative morbidity is very high, and preoperative chemotherapy is recommended.
Neuroblastoma
Neuroblastoma is the most common extracranial solid tumor in children, accounting for 10% of all childhood cancers. Approximately two thirds of patients with neuroblastoma have metastatic disease at diagnosis, typically of the bone or bone marrow, and their prognosis is very poor, although it varies according to many clinical and biologic risk factors. Pulmonary involvement at diagnosis is extremely rare, occurring in less than 1% of patients. In patients with metastatic disease, lung metastases may be present in 4% of patients, are never isolated, and are always found in the context of a very aggressive clinical and biologic behavior. On radiographs, these lesions appear as multiple, small, bilateral, noncalcified nodules.
Hepatoblastoma
Hepatoblastoma is a very rare malignancy in the overall population, but it accounts for 75% of all liver cancers in children. Lung metastases are very common; 20% of pediatric patients have pulmonary disease at diagnosis, and the lungs are the most common site of recurrence. Metastatic hepatoblastoma is curable if the tumor responds to chemotherapy and the lung metastases are resected. With this aggressive approach, the 5-year overall survival estimates are approximately 50%.
Imaging Considerations
Multiple studies have shown that even in children with tumors known to metastasize to the lungs, a significant proportion of lesions found on chest CT scans are benign. Although granulomas develop less frequently in children than in adults, several other benign conditions may develop in the lungs of children, including the growth of normal intrapulmonary lymph nodes, hamartoma, round pneumonia, atelectasis (particularly in children requiring sedation or anesthesia), and inflammatory pseudotumor. Distinguishing benign from malignant pulmonary nodules may be difficult ( Fig. 7-6 ).

The availability of helical CT has improved the quality of the chest images by diminishing the effect of breathing and cardiac motion on the readability of the scan. McCarville and associates investigated the imaging features of 81 pulmonary nodules in 41 patients with solid tumors who had undergone a thoracotomy. The new chest CT scans were able to detect 75% of the nodules found at surgery; 44% of the patients had benign lesions only. Sharply defined nodules were more likely to be malignant in children than in adults. Although node size was not always a predictor of malignancy, small (<5 mm) nodules were less likely to be malignant, particularly small solitary nodules. New technologies such as PET/CT may allow us to assess more accurately and noninvasively the metabolic activity of pulmonary nodules.
Primary Lung Neoplasms
Pleuropulmonary blastoma (PPB) is a unique dysontogenetic neoplasm of childhood that appears as a pulmonary or pleural-based mass, with a primitive, histologically variable mixed blastomatous and sarcomatous appearance. It usually manifests in the first 3 to 4 years of life with respiratory distress, fever, cough, or chest pain. Cystic changes and pneumothorax are common on CT scans. The lower lobes are more commonly involved, and pleural invasion and effusion are typical. PPB can be divided into three morphologic types or subtypes based on the cystic, cystic and solid, or solid appearance of the tumor, as determined by the gross and microscopic examination. The exclusively cystic or type I PPB is the least complex, manifests at an earlier age than the other two forms, and is more readily resectable. The rhabdomyosarcomatous component is a prominent feature of type II and type III PPB, and it is the exclusive malignant element in type I. PPB is an aggressive malignancy, and metastases to the brain are common.
Type I PPB usually has a good prognosis if complete resection is achieved. The outcome associated with type II and type III PPB is much worse, with 5-year survival rates of less than 50%. These patients need a very aggressive treatment approach, with wide resection and systemic chemotherapy. Other, less common primary malignancies of the lungs include carcinoid and primary sarcomas such as malignant peripheral nerve sheath tumor and fibrosarcoma.
Inflammatory Pseudotumor
Inflammatory pseudotumor (plasma cell granuloma, inflammatory myofibroblastic tumor) is a rare benign neoplasm consisting of histiocytes, myofibroblasts, plasma cells, and spindle-shaped mesenchymal cells. Inflammatory pseudotumor most often affects children and young adults, and it can be found in pulmonary and extrapulmonary locations, most commonly the abdomen. Despite its benign nature, it may be difficult to differentiate from a malignancy because of its local invasiveness and its tendency to recur. Pulmonary pseudotumor is often encountered as an incidental finding in a chest radiograph.
When a pseudotumor is symptomatic, patients may present with nonspecific respiratory symptoms, such as cough, shortness of breath, chest pain, hemoptysis, and clubbing. Laboratory findings consistent with inflammation are common. The etiologic factors are not clearly established, although an infectious cause is suspected. Although there are some anecdotal cases of spontaneous regression, the treatment of pulmonary inflammatory pseudotumor is surgical. Local recurrences are well described; for this reason, an aggressive surgical resection at diagnosis is always recommended.
Hematologic Disorders
Sickle Cell Disease
Sickle cell disease (SCD) comprises a group of inherited hemoglobinopathies characterized by a predominance of hemoglobin (Hb) S. Included in SCD are sickle cell anemia (Hb SS, the homozygous state for the β S globin gene), Hb SC (Hb S and Hb C), sickle β-thalassemia syndrome (either Hb β 0 thalassemia or Hb β + thalassemia in combination with Hb S), and other combinations of Hb S and abnormal hemoglobins. These hemoglobinopathies result from the substitution of valine for glutamic acid at position 6 of the β globin chain, which alters the quaternary structure of hemoglobin and reduces the solubility of deoxyhemoglobin S to approximately 10% of that of the normal deoxyhemoglobin A. After deoxygenation of the erythrocyte, Hb S undergoes intracellular polymerization. As the concentration of Hb S increases, the erythrocytes collapse into a sickle shape, either reversibly or irreversibly. Repeated cycles of polymerization induce a series of erythrocyte abnormalities, including cytoplasmic and membrane rigidity and cellular dehydration. Elevated blood viscosity and microvascular occlusion then develop. The clinical features of the sickle cell syndromes include chronic hemolytic anemia, frequent infections owing to loss of splenic function, and microvascular obstruction producing acute and chronic anemia and organ damage from infarction and fibrosis.
Patients with sickle cell anemia frequently experience acute pulmonary complications, such as asthma, thromboembolism, and acute chest syndrome (ACS). Results of several studies suggest that asthma is a significant contributing factor for acute pulmonary complications, and 35% of children with SCD have obstructive lung disease. Individuals with SCD seem to be at risk of thromboembolism because of a hypercoagulable state. The term “acute chest syndrome” reflects the difficulty of establishing a definitive cause for these acute pulmonary episodes, particularly in distinguishing infection from pulmonary infarction by microvascular occlusion ( Fig. 7-7 ).

Acute Chest Syndrome
Definition
ACS is a clinical pulmonary exacerbation of SCD manifested by fever; a new infiltrate on chest radiograph; and one or more pulmonary signs or symptoms, such as cough, dyspnea, and chest pain. As with pneumonias in children without SCD, the initial radiograph may not show a new infiltrate. The most common presenting signs are fever, cough, tachypnea, crackles, and hypoxemia. Fever is more common in children with ACS, and pain is more common in adults. ACS is the second leading reason for hospitalization (after vaso-occlusive painful crisis) and the leading cause of death in patients with SCD. Sudden death in the setting of ACS may be associated with acute exacerbations of pulmonary hypertension.
Epidemiology
Vaso-occlusive painful crisis and ACS are the most common sickle cell–related events during the first decade of life. The risk of vaso-occlusive painful crisis and ACS begins in the first year and increases steadily; ACS is experienced by half of all patients with Hb SS disease by 6 years of age. Of patients with sickle cell anemia (Hb SS) experiencing a first ACS, 25% have a recurrent event in 6 months. The risk is decreased with greater levels of fetal hemoglobin (Hb F) and is increased with the magnitude of the anemia and with elevated steady-state white blood cell counts. Patients with Hb SC disease also are at risk, although the incidence and frequency of vaso-occlusive episodes, including ACS, is lower.
Etiology
The etiology of ACS is complex and heterogeneous. In addition to the expected intravascular pulmonary thrombosis, ACS is marked by the co-occurrence of infection, fat embolism (presumably from bone infarcts), hypoventilation, edema, and reactive airway disease, and by secondary problems of mucus hypersecretion and atelectasis. Pulmonary infarction is the presumed cause in 16% of cases, and fat embolism is the presumed cause in 9%. Because multiple etiologic factors are usually involved, the diagnostic label of ACS is strongly preferred to pneumonia. A specific cause of ACS can be identified in 50% of the cases.
In children with SCD, infections are more commonly documented, as are associated bacteremias. An infectious etiology can be identified in one third of the patients; the most common agents are viruses, such as respiratory syncytial virus, parvovirus, and rhinovirus, particularly during winter months. These can be diagnosed through the immunofluorescent assessment of nasal secretions or by serial measurements of specific antibody titers. Atypical bacteria, such as Mycoplasma pneumoniae and Chlamydia pneumoniae , mostly cause bacterial infections. Pneumonia from these agents is more severe in SCD than in normal children. In the era of long-term antibiotic prophylaxis and immunizations for Streptococcus pneumoniae and Haemophilus influenzae , these bacteria are less commonly responsible for ACS than previously, and other causes such as community-acquired pneumonia have arisen. In 45% of the cases, an etiology cannot be identified.
The vasculopathy of SCD progresses in the first years of life to render the spleen incapable of performing its immune functions, including the generation of antibodies to encapsulated organisms. In addition to this “functional asplenia,” there are abnormalities of the complement defense system. Together, these deficits predispose patients, especially children, to blood-borne infections with encapsulated organisms. Antimicrobial prophylaxis with penicillin and immunization against these organisms ( S. pneumoniae and H. influenzae ) are important strategies to reduce episodes of bacterial sepsis and recurrences of ACS. After age 6 months, annual immunizations against influenza are recommended.
Clinical Presentation
Nearly half of patients with SCD are usually admitted with a diagnosis other than ACS, usually a vaso-occlusive crisis; pain is a prodrome of ACS. The clinical presentation of ACS is similar to that of pneumonia. Cardinal features comprise fever; a new infiltrate on radiograph; symptoms including cough, shortness of breath, and chest pain; signs including tachypnea, crackles, and hypoxia; and pain, including a greater frequency of chest, rib, and abdominal pain in children, and a greater frequency of extremity pain in adolescents and adults. The presenting chest radiograph may be normal, as it may be with clinical pneumonia, so a second radiograph should be obtained as indicated by clinical suspicion. Multilobar involvement, especially of the lower lobes, and effusion are present in two thirds of patients. Upper and middle lobe involvement is more common in children, and isolated lower lobe involvement is more common in adults.
Abnormalities seen on radiographs tend to progress. Oxygenation and hemoglobin decrease after diagnosis, despite aggressive intervention. Hypoxia is common, but younger patients are less likely to require oxygen.
Bronchoscopy and BAL provide adequate material, and the diagnostic yield is high, but it is associated with risk of complications. The mean hospital stay is 10 days. Adults are more likely to have complications during hospitalization; neurologic events occur in 10% of patients. The most common is altered mental status, although cerebrovascular accidents also are common.
Pathogenesis
The pathogenesis of ACS is the result of a complex series of reactions involving activation of the endothelium, often caused by an infection, and subsequent adherence of sickle erythrocytes. This sequence of events leads to a partial obstruction of microcirculatory flow; prolonged transit allows extensive polymerization of Hb S with its resultant erythrocyte rigidity. Trapping of poorly deformable sickle erythrocytes results in transient or prolonged obstruction, ischemia, and further endothelial activation. Various adhesive proteins expressed on the sickle erythrocyte membrane interact with the corresponding molecules on the endothelial cell and are mediated by plasma ligands, such as thrombospondin and von Willebrand factor. Hb S polymerization generates reactive oxygen species, which activates the transcription factor nuclear factor-κB, which upregulates expression of the adhesion molecule VCAM-1 on the endothelium, facilitating erythrocyte adhesion.
Granulocytes and monocytes also play an important role in microvascular occlusion. Additional vasoactive components also may play a role in the ACS process. Endothelin is a potent vasoconstrictor of the pulmonary vascular bed, and its levels are increased with hypoxemia. In patients with SCD, endothelin-1 levels are increased during the steady state, and increase sharply during the ACS. Nitric oxide, in addition to providing vasodilation of the pulmonary vasculature, downregulates VCAM-1 and inhibits the adherence of normal sickle erythrocytes to vascular endothelium.
Treatment
In the management of ACS, clinicians need to consider the evolving nature of the ACS. Most patients present with pain or develop pain during admission, and in many instances, chest pain may result in hypoventilation, atelectasis, or pneumonia. Incentive spirometry must be started early and be aggressively implemented. A judicious use of opioid analgesics must be implemented early in management.
Dehydration predisposes to vaso-occlusive painful crisis and ACS, and commonly follows episodes of fever, tachypnea, or anorexia. Aggressive rehydration is always indicated in the management of vaso-occlusive crises. Rehydration is often accomplished by the administration of fluids at a rate of 1.5 times maintenance, which is reduced to maintenance when the patient is normovolemic. Overhydration should be avoided because pulmonary edema and worsening of the disease process might ensue. Empiric antibiotic therapy always must include a combination of a macrolide and a second-generation cephalosporin. Vancomycin also is recommended if fever persists, and there is history of infection with penicillin-resistant S. pneumoniae , or if the patient resides in an area with a high prevalence of resistance of this organism. Oxygen supplementation also is indicated for decreases in pulse oximetry greater than 4% over baseline, or for values less than 92%.
Oxygenation significantly improves with simple blood transfusion and exchange transfusion. Early transfusions are indicated for patients at high risk of complications; patients presenting with severe anemia, thrombocytopenia, and multilobar pneumonia should receive a transfusion even before respiratory distress develops. A post-transfusion increase in hemoglobin levels greater than 11 g/dL must be avoided, however, because of the risk of sudden increase in blood viscosity that could lead to thrombosis and ischemic episodes. Exchange transfusion is indicated if the patient’s baseline hemoglobin is elevated, or the patient has more severe disease, worsening hypoxemia, neurologic abnormalities, or multiorgan failure.
Evidence is mounting that reactive airway disease may play a role as a cause and as a consequence of ACS. Reactive airway disease may occur in almost half of SCD patients, and seems to be a risk factor for the development of ACS and pulmonary hypertension. The concurrent association of reactive airway disease and ACS is associated with more severe exacerbation and prolonged hospitalization. Reactive airway disease is associated with the release of a wider variety of inflammatory mediators and cytokines than with ACS alone, and is associated with airway edema and mucus hypersecretion, factors known to worsen the course of ACS.
Treatment of reactive airway disease exacerbations in the context of ACS is similar to standard acute asthma care. There is a theoretical basis for steroid use because activation of the inflammatory cascade plays a central role in ACS; however, its role is still unclear. A short course of steroids may reduce the length of hospitalization in children and the need for analgesics and oxygen supplementation, but a rebound effect may occur. Bronchoscopy is recommended when patients do not respond to initial therapy. Aggressive mechanical ventilatory support, including extracorporeal membrane oxygenation, may be necessary; nitric oxide also may be used.
Secondary Complications
As with community-acquired pneumonias, ACS is frequently associated with atelectasis or pleural effusions or both. The pleural fluid is usually sterile and exudative, but may develop empyema. The atelectasis may or may not be associated with retained airway mucus secretions. In some cases, the mucus strings are extensive and dehydrated and may form bronchial casts. Because of their firmness, the syndrome has been referred to as plastic bronchitis. In these cases, fiberoptic bronchoscopy with washes of saline, DNase, or N -acetyl cysteine may be useful in clearing the airway obstruction and expediting the resolution of the atelectasis.
Chronic Sickle Cell Lung Disease
Chronic sickle lung disease is a progressive disorder that is insidious in onset during childhood and may be asymptomatic in the absence of frequent exacerbations of ACS. The end stage of progression is characterized by persistent hypoxemia, radiographic evidence of interstitial fibrosis, restrictive lung disease on pulmonary function testing, pulmonary hypertension, and cor pulmonale. Pulmonary hypertension in patients with SCD has been associated with worsened survival. Close pulmonary and cardiac follow-up are essential to monitor the progression of chronic sickle lung disease.
Pulmonary Hypertension
The obliterative vasculopathy of pulmonary hypertension is evident in one third of postmortem studies of patients with SCD, but the true prevalence is unknown. This disorder is most frequently recognized in adults, but almost certainly originates earlier in childhood. Although a worsened prognosis for chronic sickle lung disease and pulmonary hypertension is associated with recurrent episodes of ACS, the prevalence of pulmonary hypertension also is high in patients without a history of ACS.
This pathophysiologic predisposition is related to the combination of hemolytic anemia and “functional asplenia.” Hemolysis in combination with inadequate splenic clearance results in the accumulation of iron-rich hemoglobin, which functions as a scavenger of nitric oxide and catalyzes the formation of oxygen radical species. Arginase is simultaneously released from erythrocytes and serves to limit the supply of arginine to nitric oxide synthase, limiting the synthesis of nitric oxide. The nitric oxide serves a crucial function in promoting pulmonary vasodilation and limiting the impact of many factors contributing to the vascular remodeling of pulmonary hypertension. The functional asplenia contributes to the remodeling by the failure to clear platelet-derived mediators and cytokines that promote adherence of erythrocytes to the endothelium and cause microthrombosis.
The diagnosis of pulmonary hypertension in SCD is ominous, with a 10-fold increase in the risk for death and survival less than 50% within 4 years. The more recent validation of echocardiography as a diagnostic tool for pulmonary hypertension in SCD should promote earlier detection. Several studies have suggested the potential role for oxygen therapy, red blood cell transfusions, inhaled nitric oxide, intravenous arginine, prostacyclin, and the newer vasodilators. Controlled long-term studies are needed to determine the effectiveness of these and other therapies.
Much of the improved quality of life and survival of patients with SCD is attributed to the details of preventive health care. Because the spleen is a victim of sickle cell vasculopathy in the first few years of life (functional asplenia), functional immunity to encapsulated organisms is impaired, and the risk for sepsis from these organisms is increased. Protective strategies include penicillin prophylaxis and immunizations to S. pneumoniae and H. influenzae . Penicillin (or erythromycin) is administered twice daily from early infancy to age 5 years in children with the more severe hemoglobinopathies (Hb SS and Hb S/β 0 thalassemia) and through age 3 years in children with less severe variants of SCD, such as Hb SC. Prophylactic antibiotics are given permanently to patients who have undergone splenectomy or have experienced pneumococcal sepsis. The 7-valent pneumococcal vaccine is administered to infants beginning at 2 months of age with four doses administered by the age of 15 months, and the 23-valent vaccine is administered at ages 2 and 5 years and then every 5 to 7 years thereafter. The usual childhood schedule is followed for H. influenzae type B immunizations. Immunizations to influenza virus are administered annually after age 6 months. The usual childhood immunization schedule for hepatitis B is emphasized to prevent this potential complication of recurrent red blood cell transfusions.
Therapies to correct the basic genetic or β globin protein defect of SCD and therapies to prevent the progression of SCD do not yet exist. The major current therapeutic strategies are designed to reduce the proportion of erythrocytes carrying predominantly sickle hemoglobin (Hb S) and to delay the progression of pulmonary hypertension.
Of all the new therapies for treating SCD, hydroxyurea is the most promising. Several pediatric and adult trials have reported decreases in pain, the incidence of ACS, and overall mortality. Hydroxyurea therapy was initiated because of the drug’s ability to increase Hb F production. This increase in Hb F and a concomitant increase in red blood cell volume effectively reduce the intracellular concentration of Hb S and result in decreased sickling. Hydroxyurea has other beneficial effects, including a decrease in neutrophils and monocytes (intervening in the inflammatory cascade), an increase in red blood cell deformability, and a decrease in the adhesive receptors in the erythrocyte membrane. Trials of increasing length of hydroxyurea therapy in adults and children have shown safety and clinical efficacy, including a reduction in the incidence of ACS.
Other options include therapy with erythropoietin to increase Hb F, but its effectiveness has been suboptimal. Because of this and its substantial expense, use of erythropoietin has been limited. Its primary use is as adjunctive therapy for patients whose response to hydroxyurea is suboptimal or for patients with religious objections to blood transfusions.
Transfusion therapy of normal erythrocytes serves to depress the bone marrow production of erythrocytes expressing sickle hemoglobin. The timing of transfusion may be acute, intermittent, or chronic. Adverse effects of recurrent transfusions include iron overload, blood-borne infections, and alloimmunization, complications that can be reduced by erythrocytopheresis, highly specific crossmatching, and the newer iron-chelating agents. Current indications for red blood cell transfusion therapy in SCD include severe cases of vaso-occlusive painful crisis, recurrent or severe ACS, severe or worsening anemia, and primary and secondary stroke prevention in patients at increased risk.
Because of the potentially serious complications of bone marrow transplantation, in the United States it is offered primarily to patients who have had serious complications, such as stroke or recurrent ACS, and to patients with HLA-matched sibling donors. Its greatest benefit is, however, to patients who have neither experienced significant complications nor developed significant chronic sickle lung disease.
Efforts to develop effective gene therapy or β globin substitution have failed so far. The primary impediment has been the development of a vector suitable for the delivery of the replacement of β globin DNA to bone marrow precursors.
Histiocytic Disorders of the Lung
The histiocytoses are a group of rare hematopoietic disorders characterized by the clonal proliferation and pathologic accumulation of cells of the mononuclear-phagocyte system in tissues. The mononuclear-phagocyte system is a system of cells whose primary functions consist of phagocytosis of foreign material, antigen processing, and antigen presentation to lymphocytes. Mononuclear phagocytes are subclassified into two major classes, macrophages and dendritic cells, based on morphologic and functional characteristics. The most common pulmonary phagocytes are the alveolar and the airway macrophages. They are indistinguishable except for their location within the respiratory tract. These cells are primarily responsible for the sequestering and disposal of inhaled particles and pulmonary surfactant. They play an active role in the maintenance and remodeling of the extracellular connective tissue matrix through the secretion of cytokines that regulate matrix production and degradation. Interstitial macrophages are the second group of pulmonary macrophages with an improved antigen-presenting capability. The second class of mononuclear phagocytes, the dendritic cells, consist of dendritic cells of the lymphoid follicle and Langerhans cells of the skin and other organs.
The histiocytic disorders can be classified into three classes based on the pathologic cells present within the lesions: class I, dendritic cell histiocytosis; class II, non-dendritic cell histiocytosis, and class III, malignant histiocytosis. The rest of this discussion is dedicated to the pathophysiology, clinical presentation, diagnosis, and treatment of Langerhans cell histiocytosis (LCH) and histiocytic disorders involving the lung.
LCH is the most common of the dendritic cell histiocytoses. It is a proliferative disorder of activated Langerhans cells and is characterized by variable biologic behavior and a spectrum of distinct clinical presentations. The pathogenesis of LCH is poorly understood. Because LCH has been shown to be a monoclonal condition, it has been considered a neoplastic disorder. Different patterns of clinical involvement indicate other pathogenetic mechanisms, however. The benign histopathologic appearance of the lesions, the occurrence of spontaneous remissions, and the ability to respond to immunomodulation suggest a reactive clonal disorder, rather than a malignant process, at least in some cases. Langerhans cells, similar to other dendritic cells, have a crucial role in the immune system, and it has been suggested that LCH could be the result of immune dysregulation. Although no consistent immunologic abnormalities have been described, there is increasing evidence that LCH may be the result of an uncontrolled and abnormal proliferation of Langerhans cells secondary to immune dysregulation or after exposure to an as-yet-undetermined stimulus.
The diagnosis of LCH requires a biopsy with the specimen showing typical pathologic features of Langerhans cell infiltrates and confirmatory positive staining for CD1a and S-100. Alternatively, the demonstration of Birbeck granules by electron microscopy has been used for confirmation of this diagnosis. The diagnosis of pulmonary LCH in children with multisystem disease is usually clinical because the histologic diagnosis is usually made from extrapulmonary lesions. In the rare cases of isolated lung disease, a morphologic confirmation is required, and it can be obtained by lung biopsy or by BAL. In the latter case, the presence of greater than 5% of CD1a-positive cells in the BAL fluid is very suggestive of LCH. BAL also is a good tool for monitoring response and follow-up.
The histopathology of the lesion is uniform, regardless of the clinical severity of the disease. It consists of collections of Langerhans cells, interdigitating cells, and macrophages, accompanied by T lymphocytes with variable numbers of multinucleated giant histiocytes and eosinophils.
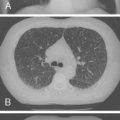
In light of the pathologic basis of the diagnosis of LCH and the variable nature of clinical presentations, LCH is now subclassified by the degree of organ involvement: localized single-system disease, multifocal single-system disease, and multisystem disease. Localized LCH, involving skin, lymph node, or bones, usually has a good prognosis. The most common presentation of multifocal LCH includes diffuse skin involvement and involvement of multiple bones. The oral cavity; lymph nodes; and, to a lesser extent, lungs, liver, and brain are common sites of involvement in this disease. Lung involvement has been reported to occur in 20% to 50% of patients with multisystem disease. Radiographic changes consistent with past lung involvement are present in 15% to 20% of long-term survivors ( Fig. 7-8A ).

Related posts:
 Chronic Lung Disease of Infancy
Chronic Lung Disease of Infancy
 Pulmonary Manifestations of Human Immunodeficiency Virus (HIV) Infection
Pulmonary Manifestations of Human Immunodeficiency Virus (HIV) Infection
 Pulmonary Manifestations of Endocrine and Metabolic Diseases
Pulmonary Manifestations of Endocrine and Metabolic Diseases
 Pulmonary Manifestations of Rheumatoid Diseases
Pulmonary Manifestations of Rheumatoid Diseases
 Pulmonary Manifestations of Gastrointestinal Diseases
Pulmonary Manifestations of Gastrointestinal Diseases
 Pulmonary Manifestations of Dermatologic Diseases
Pulmonary Manifestations of Dermatologic Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


