The pulmonary vasculitides are a heterogeneous group of primary or secondary diseases, in which there is inflammation that may lead to progressive destruction of the pulmonary microvasculature. Pulmonary involvement may develop because the lung has an extensive vascular and microvascular network, sensitizing antigens can easily reach the lung, and there are large numbers of vasoactive and activated immune cells in the lung. Primary or isolated pulmonary vasculitis is extremely rare in children. Pulmonary vasculitis is an unusual condition in children and is almost always seen in conjunction with a systemic vasculitis syndrome. This chapter reviews common systemic vasculitides associated with pulmonary disease and discusses presentation, diagnosis, and therapy.
Definition of Vasculitis
Before embarking on a discussion of specific illnesses associated with inflammation of blood vessels and pulmonary involvement, clarifying what is meant by “vasculitis,” how vasculitides may manifest systemically, and some of the serologic markers associated with these diseases is in order. Simply defined, vasculitis is inflammation of the walls of blood vessels. Perivascular cuffing is not a vasculitis. The involved vessels may be arteries, veins, or capillaries; large, medium-sized, or small arteries may be involved; the infiltrate may be neutrophils, lymphocytes, eosinophils, or plasma cells; the inflammation may be necrotizing or granulomatous; there may or may not be immune complex deposition or antineutrophil antibodies. All of these characteristics determine the specific type of vasculitis.
A definite diagnosis cannot be made without tissue biopsy, although several clinical syndromes are fairly typical and should suggest what type of pathology one is likely to see. The clinical and pathologic features vary, and depend on the site and type of blood vessel involved. Although many vasculitides affect adults and children, some, such as Kawasaki disease, occur almost exclusively in children. Other vasculitides, such as temporal arteritis, rarely, if ever, occur in childhood, and others, such as polyarteritis and Wegener granulomatosis, have different etiologic, clinical, and prognostic characteristics in children. It was thought inappropriate to continue applying the adult classification criteria to vasculitis occurring in childhood. The newly adopted classification for childhood vasculitides comes from the consensus criteria endorsed by the European League Against Rheumatism and the Pediatric Rheumatology European Society ( Table 11-1 ).
|
Despite extensive investigation, the pathogenesis of vascular inflammation is unknown in most cases. Vasculitis probably results from the combination of multiple risk factors, including genetic predisposition, environmental factors (infection and inhalation of particulate matter), and chance. Figure 11-1 shows the distribution of vasculitis type in vessels of various sizes. There is a great deal of overlap regarding which vessels are involved in some common vasculitides, notably Wegener granulomatosis and microscopic polyangiitis. Large vessel vasculitides, such as giant cell arteritis and Takayasu arteritis, do not cause much pulmonary disease because the intrapulmonary vessels are medium-sized arteries or smaller. Medium-sized vessel arteritides include polyarteritis nodosa. Classic polyarteritis nodosa is uncommon in children and generally not associated with lung disease, although case reports of lung disease in association with polyarteritis nodosa and Kawasaki disease have been published. The small vessel variant of polyarteritis nodosa, microscopic polyangiitis, is associated with lung disease and is discussed in detail subsequently.

Typically, patients with small vessel vasculitides present with pulmonary manifestations. These include Wegener granulomatosis, microscopic polyangiitis, Henoch-Schönlein purpura, Churg-Strauss syndrome, and Goodpasture syndrome.
Systemic manifestations of vasculitides include malaise, fever, weight loss, joint pain, kidney disease, and skin rash. Diagnosis of a specific vasculitis syndrome depends on clinical and serologic characteristics.
Typical vasculitis syndromes are associated with deposition of immune complexes in blood vessel walls. Goodpasture syndrome is the classic example of IgA-related complex deposition. A subset of small vessel vasculitides has minimal immune-complex deposition in vessel walls. These so-called pauci-immune vasculitides include Wegener granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome, and renal-limited vasculitis. Small amounts of immune complexes form on the surfaces of blood vessels in these diseases, but differ from larger vessel vasculitides in that there is not the gross accumulation of immune complexes seen in the large vessel diseases; also, the small vessel vasculitides are associated with a greater degree of necrotizing injury. The precise cause of tissue injury in Wegener granulomatosis and microscopic polyangiitis is uncertain, but there is a strong association between disease activity and the presence of antineutrophil cytoplasmic antibodies (ANCA).
There is mounting evidence that ANCA play a causative role in Wegener granulomatosis and microscopic polyangiitis ( Fig. 11-2 ). A strong case can be made that the presence of ANCA directly leads to disease based on several lines of evidence. First, an infant who was born to a mother with high circulating levels of ANCA developed pulmonary hemorrhage and glomerulonephritis within days of birth. The infant’s serum was ANCA-positive. Treatment with corticosteroids and exchange transfusion brought the disease into remission. Second, neutrophils that have been activated by ANCA-IgG kill cultured endothelial cells. Third, monocytes and neutrophils interact with ANCA, and monocyte-derived mediators of inflammation can damage vessel walls further. Finally, glomerulonephritis has been precipitated in several murine models with transfer of ANCA-IgG. Taken together, these findings indicate that the presence of ANCA may be toxic to blood vessels and precipitate disease.

ANCA come in two forms, and the nomenclature has become confused as more has been learned about these antibodies. Initially, the designations used for ANCA-IgG were P-ANCA (for perinuclear staining) and C-ANCA (for cytoplasmic staining) when anti-ANCA antibodies were used to detect the presence and location of intracellular ANCA ( Fig. 11-3 ). These older terms have been replaced by more precise terms based on the specific antigens against which ANCA is directed. The target for P-ANCA is most frequently myeloperoxidase (MPO) (although some other antigens have been reported), and the target for C-ANCA is proteinase-3 (PR3). On activation, neutrophil cytoplasmic granules move to the cell surface where expression of MPO and PR3 antigens allows association with circulating ANCA. ANCA activation of neutrophils requires antigen binding and Fc-receptor engagement. Levels of PR3 antigen expression may be genetically determined, making some individuals more prone to ANCA-associated diseases than others.


Consistent with the association of these two ANCA with different disease processes, internalization of MPO-ANCA and PR3-ANCA causes different cellular responses. MPO-ANCA causes generation of intracellular oxidants, whereas PR3-ANCA causes endothelial cell apoptosis. MPO-ANCA and PR3-ANCA activate neutrophils to release mediators of acute inflammation.
Indirect immunofluorescence and enzyme-linked immunosorbent assay tests for ANCA are available. It is recommended that a combination of these tests be used if the diagnosis of an ANCA-associated vasculitis is suspected. Positive ANCA by indirect immunofluorescence alone is not specific for systemic vasculitis; a positive indirect immunofluorescence test can be seen in patients with systemic lupus erythematosus, rheumatoid arthritis, Felty syndrome, ulcerative colitis, and other multisystem conditions. It is imperative also to employ the enzyme-linked immunosorbent assay test, which differentiates PR3-ANCA from MPO-ANCA. This distinction is important because, as noted before, different ANCA may be associated with various disease processes. ANCA-associated diseases are the same in children as in adults.
Specific Vasculitis Syndromes
Pulmonary-Renal Syndromes
Numerous disorders affect the small vessels of the lungs and the kidneys. These conditions are associated most often with a combination of pulmonary hemorrhage and nephritis. Pulmonary hemorrhage may not be obvious, and can manifest simply as diffuse, patchy pulmonary infiltrates on a chest radiograph in a child who has unexplained anemia. Although easy to diagnose when the lungs and kidneys are affected simultaneously, in all of these syndromes there may be temporal separation of specific organ involvement.
The pulmonary-renal syndromes consist of Henoch-Schönlein purpura, Wegener granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome, Goodpasture syndrome, and systemic lupus erythematosus. All of these entities are associated with an inflammatory component, and children affected by them generally have an elevated erythrocyte sedimentation rate and other serum markers of inflammation. Table 11-2 highlights the clinical and laboratory features of these syndromes. The pulmonary manifestations of systemic lupus erythematosus are discussed in detail in Chapter 10 and are not reiterated here.
| HSP | GPS | WG | MPA | SLE | |
|---|---|---|---|---|---|
| Pulmonary hemorrhage | 0 to + | ++++ | +++ | +++ | + to ++ |
| Glomerulonephritis | ++++ | ++++ | ++++ | ++++ | +++ to ++++ |
| Upper airway involvement | 0 | 0 | ++++ | ++ | + to ++ |
| Skin rash | ++++ | 0 to + | +++ | +++ | ++++ |
| Arthralgia | ++++ | 0 | +++ | +++ | ++++ |
| Elevated ESR | + | 0 | ++++ | ++++ | ++++ |
| Abdominal involvement | ++++ | 0 | 0 | 0 | 0 |
| Serology | IgA positive, IgM positive | Anti-GBM (rarely P-ANCA) | C-ANCA (rarely P-ANCA) | P-ANCA, C-ANCA | ANA, anti–double-stranded DNA (rarely P-ANCA) |
* 0 = not present; increasing association with increasing number of + signs.
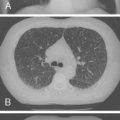
All of the pulmonary-renal syndromes occur with a low frequency in childhood. The most common small vessel vasculitis in childhood, Henoch-Schönlein purpura, has the lowest incidence of associated pulmonary disease, and all the other forms of vasculitis in childhood are quite rare in general, so despite the fact that they are commonly associated with lung disease, they are still unusual causes of pulmonary hemorrhage. When a pulmonary-renal syndrome does occur, however, it can be devastating. Pulmonary hemorrhage can be massive and life-threatening, and usually manifests as anemia in association with pulmonary infiltrates on chest radiographs ( Fig. 11-4 ). Glomerulonephritis can be sudden and severe with little prodrome. Early treatment with anti-inflammatory agents is lifesaving, so a high degree of suspicion for and rapid recognition of this syndrome are key.

Any child with anemia and pulmonary infiltrates must be suspected of having pulmonary hemorrhage, be it idiopathic, traumatic, or due to small vessel vasculitis. The skin also should be examined for signs of cutaneous vasculitis (palpable purpura, erythema nodosum, ulceration, splinter hemorrhages), and the nose, ears, and nasopharynx and oropharynx should be examined for the presence of bleeding or granuloma. When the diagnosis of small vessel vasculitis is being considered, urine sediment must be evaluated for the presence of cellular casts and protein. Serum creatinine levels can be maintained in the normal range even in the face of glomerular injury, so waiting for an increase in serum urea and creatinine unduly delays the diagnosis.
Henoch-Schönlein Purpura
Henoch-Schönlein purpura is the most common form of small vessel vasculitis in childhood. Its incidence is estimated to be 10 to 14 per 100,000, with boys affected twice as frequently as girls. In the United States, the prevalence peaks in children 5 years old. Approximately 75% of cases occur in children 2 to 11 years old. Henoch-Schönlein purpura is rare in infants and young children.
The etiology of Henoch-Schönlein purpura is largely unknown, although it commonly follows an upper respiratory tract infection. Group A streptococcus has been implicated as a trigger for Henoch-Schönlein purpura because many patients with Henoch-Schönlein purpura have elevated antistreptolysin O antibodies. The pathophysiology of Henoch-Schönlein purpura is not fully understood, but IgA plays a crucial role in the immunopathogenesis, as evidenced by increased serum IgA concentrations, IgA-containing circulating immune complexes, and IgA immune complex deposition in vessel walls and renal mesangium. Henoch-Schönlein purpura is almost exclusively associated with abnormalities involving IgA1, rather than IgA2. IgA-negative biopsies occur in 10% to 25% of cases of Henoch-Schönlein purpura, although it is possible that these IgA-negative cases represent other, unidentified vasculitic syndromes misdiagnosed as Henoch-Schönlein purpura because of overlapping features.
The classic presentation includes erythematous papules followed by nonthrombocytopenic palpable purpura in the lower extremities, trunk, and face; arthralgia or arthritis; abdominal pain; gastrointestinal bleeding; and nephritis. Renal involvement occurs in approximately 60% of cases and leads to a significant degree of morbidity ( Table 11-3 ).
| At least one of the following four conditions should be present |
| Diffuse abdominal pain |
| Any biopsy specimen showing predominant IgA deposition |
| Arthritis or arthralgia |
| Renal involvement (any hematuria or proteinuria or both) |
| In the presence of palpable purpura ( mandatory criterion ) |
Although renal abnormalities in Henoch-Schönlein purpura are common, the classic pulmonary manifestations, such as diffuse alveolar hemorrhage and interstitial pneumonitis, are thought to be infrequent. Subclinical pulmonary manifestations, including diffusion defects and radiographic anomalies, seem to be quite frequent in patients with Henoch-Schönlein purpura, but are not commonly reported. Other respiratory manifestations include pleural effusion and chylothorax. Diffuse alveolar hemorrhage is the most frequent pulmonary complication associated with Henoch-Schönlein purpura, but, as previously noted, pulmonary hemorrhage is rare in these patients, although it has been reported.
Pulmonary hemorrhage is secondary to disruption of the alveolar-capillary membranes by circulating immune complexes. In a series reported from the Mayo Clinic of 28 patients with pulmonary involvement in Henoch-Schönlein purpura, 18 patients were 20 years old or younger. Of these patients, 26 had diffuse alveolar hemorrhage, 5 of whom had associated pleural effusions; 9 patients died (3 children). The youngest reported patient with pulmonary disease associated with Henoch-Schönlein purpura was a 5-month-old infant who died of complications associated with diffuse alveolar hemorrhage and interstitial fibrosis.
The clinical presentation of patients with Henoch-Schönlein purpura complicated by diffuse alveolar hemorrhage may vary. Typically, these patients are diagnosed with Henoch-Schönlein purpura before the onset of pulmonary symptoms. Some patients may develop milder pulmonary symptoms, such as cough, which may be associated with blood-tinged sputum and crackles on lung auscultation. Other patients progress to dyspnea at rest, hemoptysis, and occasionally respiratory failure requiring intubation and ventilatory support. Blood loss into the lungs may be severe enough to require transfusion therapy. Chest radiographs show diffuse pulmonary infiltrates secondary to alveolar hemorrhage. Bronchoscopy is useful only if uncertainty exists regarding pulmonary hemorrhage versus infection as the cause of the physical examination and radiologic abnormalities. Lung biopsy is rarely needed because affected tissue generally can be obtained from less dangerous biopsy sites (skin or kidney).
Treatment of Henoch-Schönlein purpura almost always includes the use of corticosteroids. Prednisone, 1 to 2 mg/kg/day, is the initial choice. Cyclophosphamide, 2 to 3 mg/kg/day, commonly is added in cases involving severe pulmonary hemorrhage. Length of therapy depends on the time to resolution of symptoms and radiographic clearance of pulmonary infiltrates. Supportive care, including mechanical ventilation, is often necessary in cases with severe alveolar damage.
Although clinically important pulmonary disease is rare in Henoch-Schönlein purpura, even patients without respiratory symptoms have objective signs of mild lung abnormalities. In a cohort of children with Henoch-Schönlein purpura free of clinical pulmonary symptoms, patients were noted to have an impaired diffusing capacity for carbon monoxide. In this study, 70% of the patients had radiographic evidence for mild interstitial lung changes. In another group of children with Henoch-Schönlein purpura who had no evidence for lung disease clinically or radiographically, the finding of an abnormal diffusing capacity for carbon monoxide was again seen.
Wegener Granulomatosis
Wegener granulomatosis is a necrotizing granulomatous vasculitis with a broad spectrum of findings and progression. Wegener granulomatosis and microscopic polyangiitis are extremely similar diseases with great overlap in clinical and pathologic presentations. As such, it has been suggested now to combine these diseases under the rubric, “ANCA-associated small vessel vasculitis.” These two entities are discussed separately here partly for historical reasons and partly because there remain many clinical studies that focus on either one or the other of these entities. Practically, it might be impossible to distinguish Wegener granulomatosis from microscopic polyangiitis, however, even with tissue biopsy. What looks to be microscopic polyangiitis initially may prove to be Wegener granulomatosis later, when manifestations in other organs become apparent, and granulomas are present. The overlap in serology between these two entities is highlighted in Table 11-3 .
Wegener granulomatosis is uncommon in children; its peak incidence is in adults in their 60s and 70s. The etiology of Wegener granulomatosis is unknown, and it does not have specific genetic markers. In adults, systemic Wegener granulomatosis is noted for its severity and high mortality. Still, Wegener granulomatosis has been reported in children; as in adults, it generally manifests with the triad of upper airway, lower airway, and renal involvement. Children frequently present with cough, sinus pain, fever, arthralgias or arthritis, hemoptysis, and skin rashes or ulceration. Cutaneous manifestations may precede other systemic symptoms by months and occur most commonly on the legs. The predominant skin finding is palpable purpura, but lesions may be vesicular, papular, or nodular, and even urticaria has been reported. Upper airway manifestations of Wegener granulomatosis include rhinorrhea, purulent or bloody discharge, persistent otitis media, sinusitis, saddle-nose deformity, subglottic stenosis, and tracheal stenosis. Renal involvement may occur early or late in the course, and when present, it requires aggressive management to avoid rapid progression to end-stage renal disease.
The American College of Rheumatology criteria require the presence of two of the following four criteria for the diagnosis of Wegener granulomatosis: nasal-oral inflammation, abnormal findings on chest radiograph, abnormal urinalysis, and granulomatous inflammation on biopsy specimen. The presence of subglottic, tracheal, or endobronchial stenosis, and the presence of a high titer of PR3 or positive C-ANCA staining have been included more recently in the diagnostic criteria. The new classification of a child as having Wegener granulomatosis requires the presence of three of the newly established criteria of six ( Table 11-4 ).
Related posts:
 Chronic Lung Disease of Infancy
Chronic Lung Disease of Infancy
 Pulmonary Manifestations of Human Immunodeficiency Virus (HIV) Infection
Pulmonary Manifestations of Human Immunodeficiency Virus (HIV) Infection
 Pulmonary Manifestations of Endocrine and Metabolic Diseases
Pulmonary Manifestations of Endocrine and Metabolic Diseases
 Pulmonary Manifestations of Rheumatoid Diseases
Pulmonary Manifestations of Rheumatoid Diseases
 Pulmonary Manifestations of Gastrointestinal Diseases
Pulmonary Manifestations of Gastrointestinal Diseases
 Pulmonary Manifestations of Dermatologic Diseases
Pulmonary Manifestations of Dermatologic Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


