Chapter 14 Pulmonary Vascular Abnormalities
PULMONARY ARTERY HYPERTENSION
Causes
PAH may results from one of three basic mechanisms: increased flow of blood through the pulmonary vessels, decreased cross-sectional area of the pulmonary vasculature, and increased resistance to pulmonary venous drainage. These mechanisms provide a convenient framework for categorizing and understanding the large number of causes of PAH (Box 14-1). Another common way to categorize causes of PAH is to broadly divide them into precapillary causes (i.e., entities that result in increased blood flow or decreased cross-sectional area of the pulmonary vasculature) and postcapillary causes (i.e., entities that result in increased resistance to pulmonary venous drainage).
Box 14-1 Causes of Pulmonary Artery Hypertension
INCREASED PULMONARY BLOOD FLOW
Left-to-right shunts (atrial septal defect, patent ductus arteriosus, ventricular septal defect)
Increased total blood volume (thyrotoxicosis, anemia, pregnancy)
DECREASED CROSS-SECTIONAL AREA OF THE PULMONARY VASCULATURE
Radiographic Findings
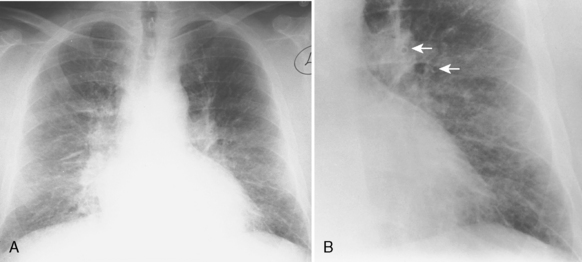
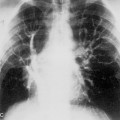
Despite the wide variety of causes of PAH, the salient radiologic features are similar. There is usually marked enlargement of the main and hilar pulmonary arteries, which rapidly taper as they course distally (Fig. 14-1). The most striking feature of PAH is the disparity in size between the central and peripheral pulmonary arteries. Right-sided ventricular cardiac enlargement may occur and is best demonstrated on the lateral chest radiograph.
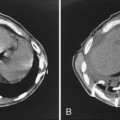
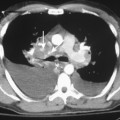
On chest radiographs, enlargement of the main pulmonary artery results in a prominent convex contour. However, the degree of pulmonary artery enlargement varies considerably among patients and conditions. A patient may have significant PAH despite a normal chest radiograph. Compared with chest radiographs, CT scans allow a more accurate determination of the size of the main pulmonary artery, and a diameter greater than 3 cm on CT usually is considered abnormal (Fig. 14-2).
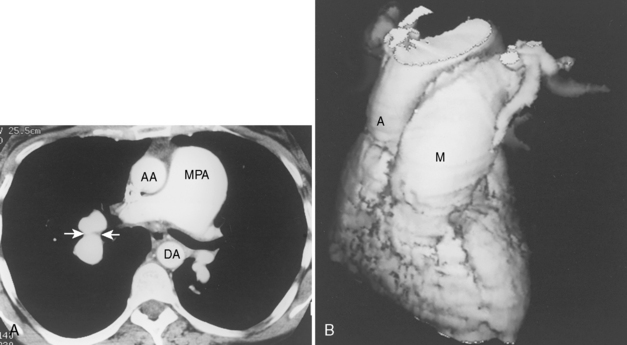
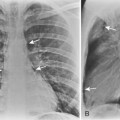
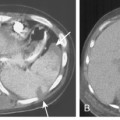
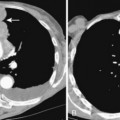
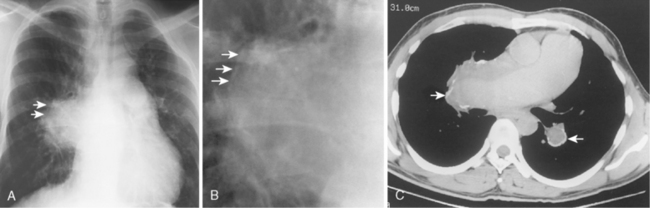
In the setting of long-standing, severe PAH, the enlarged central pulmonary arteries may develop peripheral, atherosclerotic calcification. This is an unusual finding and is most frequently seen in patients with PAH caused by Eisenmenger’s syndrome (Fig. 14-3), a condition characterized by a reversal in the direction of a long-standing, severe left-to-right shunt.
CONGESTIVE HEART FAILURE
We focus on cardiogenic pulmonary edema in this chapter. The most common cause of elevated pulmonary microvascular pressure is elevation of pulmonary venous pressure caused by diseases of the left side of the heart. Examples include left ventricular failure, diseases of the mitral valve, and left atrial abnormalities (see Box 14-1).
Interstitial Edema
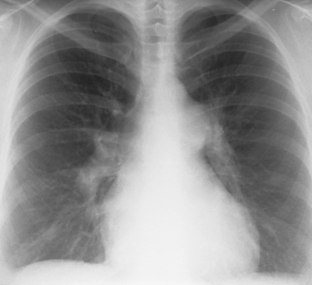
Pulmonary edema usually follows a typical course. It begins in the interstitial compartment of the lung and extends into the alveolar compartment as it increases in severity. The first phase of pulmonary edema involves the interstitial compartment. It contains two major components: the peribronchovascular sheath and the interlobular septa. Fluid within the peribronchovascular sheath results in indistinctness of the pulmonary vessels and peribronchial cuffing (Fig. 14-4). Fluid within the interlobular septa results in Kerley lines (Table 14-1 and Fig. 14-5).