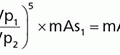1. Particle accelerators (element transmutation by absorption of a charged particle)
2. Nuclear reactor (fission product extraction or induced activity by neutron activation)
3. Radionuclide generators (device that includes the parent radionuclide from which the daughter radionuclide is extracted) (Table 16-1)
TABLE 16-1 COMPARISON OF RADIONUCLIDE PRODUCTION METHODS | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||
1. Charged particles must be accelerated to high kinetic energies to overcome and penetrate the repulsive coulombic barrier of the target atoms’ nuclei.
2. A cyclotron has a vacuum chamber between the poles of an electromagnet. Inside the vacuum chamber is a pair of hollow, semicircular electrodes, each shaped like the letter D and thus referred to as “dees.” An alternating high voltage is applied between the two dees.
3. When positive ions are injected into the center of the cyclotron, they are attracted to and accelerated toward the negatively charged dee. The static magnetic field constrains the ions to travel in a circular path, whereby the radius of the circle increases as the ions gain kinetic energy (Fig. 16-1).
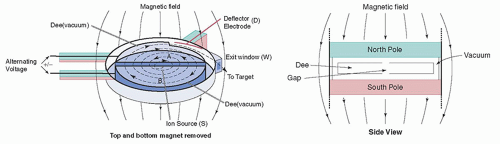
▪ FIGURE 16-1 Schematic view of a cyclotron. F. 16-1
F. 16-1
4. Halfway around the circle, the ions approach the gap between the dees; at this time, the polarity of the electrical field between the two dees is reversed, causing the ions to be accelerated toward the negative dee. This cycle is repeated again and again, with the particles accelerated each time they cross the gap, acquiring additional kinetic energy and sweeping out larger and larger circles. The cyclic nature of these events led to the name “cyclotron.”
5. The accelerated ions collide with the target nuclei, causing nuclear reactions. An incident particle may leave the target nucleus after interacting, transferring some of its energy to it, or it may be completely absorbed.
6. The specific reaction depends on the type and energy of the bombarding particle as well as the composition of the target. In either case, target nuclei are left in an excited state, and this excitation energy is disposed of through the emission of particulate (protons and neutrons) and electromagnetic (γ-rays) radiations. Depending on the design of the cyclotron, particle energies can range from a few million electron volts (MeV) to several hundred MeV.
7. In the first example below, an accelerated proton (p) is absorbed by a stable atom of zinc-68 (68Zn) after which two neutrons are emitted, leaving an atom of gallium-67 as the reaction product.
Example reactions of industrial cyclotron-produced radionuclides used in nuclear medicine imaging:
RADIONUCLIDE
CHARGED-PARTICLE PRODUCTION REACTION
Gallium-67
68Zn (p, 2n)67Ga
Iodine-123
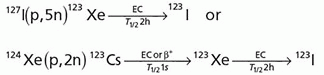
Indium-111
109Ag (α, 2n)111In or 111Cd (p, n)111In or 112Cd (p, 2n)111In
Cobalt-57
56Fe (d, n)57Co
Thallium-201
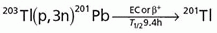
8. Industrial cyclotron facilities that produce large activities of clinically useful radionuclides are very expensive and require substantial cyclotron and radiochemistry support staff and facilities. Cyclotron-produced radionuclides are usually more expensive than those produced by other technologies.
9. Much smaller, specialized cyclotrons, installed in commercial radiopharmacies serving metropolitan areas or in hospitals, produce positron-emitting radionuclides for positron emission tomography (PET).
10. These cyclotrons operate at lower energies (10 to 30 MeV) than industrial cyclotrons and commonly accelerate H– ions, which is a proton with two orbital electrons. The beam is extracted by passing it through a carbon stripping foil, which removes the electrons thus creating an H+ ion (proton) beam.
11. The medical cyclotrons are usually located near the PET imaging system because of the short half-lives of the radionuclides produced. In the interests of design simplicity and cost, some medical cyclotrons accelerate only protons. These advantages may be offset for particular productions such as 15O when an expensive rare isotope 15N that requires proton bombardment must be used in place of the cheap and abundant 14N isotope that requires deuteron bombardment.
12. Fluorine-18 (F-18) is an exception to this generalization owing to its longer half-life (110 min). Regional production and distribution of 18F is thus an option for this commonly used PET radionuclide.
Example reactions of medical center cyclotron-produced radionuclides used in nuclear medicine imaging:
RADIONUCLIDE
CHARGED PARTICLE PRODUCTION
REACTIONS/RADIONUCLIDE PHYSICAL HALF-LIFE
Fluorine-18
18O (p, n)18F
(T1/2 = 110 min)
Nitrogen-13
16O(p, α)13N
(T1/2 = 10 min)
Oxygen-15
14N(d, n)15O or 15N (p, n)15O
(T1/2 = 2.0 min)
Carbon-11
14N (p, α)11C
(T1/2 = 20.4 min)
1. Fission is the splitting of an atomic nucleus into two smaller nuclei. Whereas some unstable nuclei fission spontaneously, others require the input of energy to overcome the nuclear binding forces. This energy is often provided by the absorption of neutrons.
2. Neutrons can induce fission only in certain very heavy nuclei. Whereas high-energy neutrons can induce fission in several such nuclei, there are only three nuclei of reasonably long half-life that are fissionable by neutrons of all energies; these are called fissile nuclides.
3. The most widely used fissile nuclide is uranium-235 (U-235). Elemental uranium exists in nature primarily as U-238 (99.3%) with a small fraction of U-235 (0.7%). U-235 has a high fission cross section (i.e., high fission probability); therefore, its concentration is usually enriched (typically to 3% to 5%) to make the fuel used in nuclear reactors.
4. When a U-235 nucleus absorbs a neutron, the resulting nucleus (U-236) is in an extremely unstable excited energy state that usually promptly fissions into two smaller nuclei called fission fragments. The fission fragments separate with very high kinetic energies, with the simultaneous emission of γ radiation and the ejection of two to five neutrons per fission:

5. The fission of uranium creates fission fragment nuclei having a wide range of mass numbers. More than 200 radionuclides with mass numbers between 70 and 160 are produced (Fig. 16-2). These fission products are neutron-rich, and therefore, almost all of them decay by beta-minus (β-) particle emission.
6. The energy released by the nuclear fission of a uranium atom is more than 200 MeV. Under the right conditions, this reaction can be perpetuated if the fission neutrons interact with other U-235 atoms, causing additional fissions and leading to a self-sustaining nuclear chain reaction (Fig. 16-3).

▪ FIGURE 16-2 Fission yield as a percentage of total fission products from uranium-235. F. 16-4
F. 16-4

▪ FIGURE 16-3 Schematic of a nuclear chain reaction. The neutrons (small black circles) are not drawn to scale with respect to the uranium atoms. F. 16-5
F. 16-5
7. The probability of fission with U-235 is greatly enhanced as neutrons slow down or thermalize. The neutrons emitted from fission are very energetic (called fast neutrons) and are slowed (moderated) to thermal energies (approximately 0.025 eV) as they scatter in water in the reactor core. Good moderators are low-Z materials that slow the neutrons without absorbing a significant fraction of them. Water is the most commonly used moderator.
1. The fission products most often used in nuclear medicine are molybdenum-99 (Mo-99), iodine-131 (I-131), and xenon-133 (Xe-133). These products can be chemically separated from other fission products with essentially no stable isotopes (carrier) of the radionuclide present.
2. The concentration or specific activity (measured in MBq or Ci per gram) of these “carrier-free” fission-produced radionuclides is very high. High-specific-activity, carrier-free nuclides are preferred in radiopharmaceutical preparations to increase the labeling efficiency and minimize the mass and volume of the injected material.
1. Neutrons produced by the fission of uranium in a nuclear reactor can be used to create radionuclides by bombarding stable target material placed in the reactor. Ports exist in the reactor core between the fuel elements where samples to be irradiated are inserted. This process, called neutron activation, involves the capture of neutrons by stable nuclei, which results in the production of radioactive nuclei.
2. The most common neutron capture reaction for thermal (slow) neutrons is the (n,γ) reaction, in which the capture of the neutron by a nucleus is immediately followed by the emission of a γ-ray.
3. Other thermal neutron capture reactions include the (n,p) and (n,α) reactions, in which the neutron capture is followed by the emission of a proton or an alpha particle, respectively. Almost all radionuclides produced by neutron activation decay by beta-minus particle emission.
Example neutron activation methods of producing radionuclides used in nuclear medicine imaging:
RADIONUCLIDE
NEUTRON ACTIVATION PROCESS/RADIONUCLIDE PHYSICAL HALF-LIFE
Phosphorus-32
31P(n, γ)32P
(T1/2 = 14.3 days)
Chrominum-51
50Cr(n, γ)51Cr
(T1/2 = 27.8 days)
Iodine-125
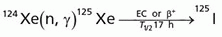
(T1/2 = 60.2 days)
1. In a molybdenum-99/technetium-99m radionuclide generator, Mo-99 (produced by nuclear fission of U-235 to yield a high-specific-activity, carrier-free parent) is loaded, in the form of ammonium molybdate (NH4+) (MoO4–), onto a porous column containing 5 to 10 g of an alumina (Al2O3) resin.
2. As with all radionuclide generators, the chemical properties of the parent and daughter are different. In the Mo-99/Tc-99m or “moly” generator, the Tc-99m is much less tightly bound than the Mo-99. The daughter is removed (eluted) by the flow of isotonic saline (the “eluant”) through the column.
3. When the saline solution is passed through the column, the chloride ions easily exchange with the TcO4– (but not the MoO4–) ions, producing sodium pertechnetate, Na+(99mTcO4–). Technetium-99m pertechnetate (99mTcO4–) is produced in a sterile, pyrogen-free form with high specific activity and a pH (approximately 5.5) that is ideally suited for radiopharmaceutical preparations.
4. Commercially, moly generators have a large reservoir of oxygenated saline (the eluant) connected by tubing to one end of the column and a vacuum extraction vial to the other. On insertion of the vacuum collection vial (contained in a shielded elution tool), saline is drawn through the column and the eluate is collected during elution, which takes about 1 to 2 min (Fig. 16-4).
5. Moly generators are typically delivered with approximately 37 to 740 GBq (1 to 20 Ci) of Mo-99, depending on the workload of the department. The larger activity generators are typically used by commercial radiopharmacies supplying radiopharmaceuticals to multiple nuclear medicine departments. The generators are shielded by the manufacture with lead, tungsten, or, in the case of higher activity, depleted uranium.
6. The activity of the daughter at the time of elution depends on the following:
a. The activity of the parent
b. The rate of formation of the daughter, which is equal to the rate of decay of the parent
c. The decay rate of the daughter
d. The time since the last elution
e. The elution efficiency (typically 80% to 90%)
1. Between elutions, the daughter (Tc-99m) builds up or “grows in” as the parent (Mo-99) continues to decay. After approximately 23 h, the Tc-99m activity reaches a maximum, at which time the production rate and the decay rate are equal and the parent and daughter are said to be in transient equilibrium. Once transient equilibrium has been achieved, the daughter activity decreases, with an apparent half-life equal to the half-life of the parent.
2. Transient equilibrium occurs when the half-life of the parent is greater than that of the daughter by a factor of approximately 10. In the general case of transient equilibrium, the daughter activity will exceed the parent at equilibrium.
3. If all of the (Mo-99) decayed to Tc-99m, the Tc-99m activity would slightly exceed (approximately 10% higher) that of the parent at equilibrium. However, approximately 12% of Mo-99 decays directly to Tc-99 without first producing Tc-99m (Fig. 16-5). Therefore, at equilibrium, the Tc-99m activity will be only approximately 97% (1.1 × 0.88) that of the parent (Mo-99) activity.
4. Moly generators (sometimes called “cows”) are usually delivered weekly and eluted (a process referred to as “milking the cow”) each morning, allowing maximal yield of the daughter. The elution process is approximately 90% efficient. This fact, together with the limitations on Tc-99m yield in the Mo-99 decay scheme, results in a maximum elution yield of approximately 85% of the Mo-99 activity at the time of elution.

▪ FIGURE 16-4 A cross-sectional diagram of a radionuclide generator interior and column detail. F. 16-7
F. 16-7

▪ FIGURE 16-5 Simplified decay scheme of Mo-99. Mo-99 decays to Tc-99m approximately 88% of the time. The balance (12%) of Mo-99 decays occurs by other beta transitions to excited states that ultimately yield Tc-99 bypassing the metastable form of technetium (Tc-99m). F. 16-8
F. 16-8
5. Therefore, a typical elution on Monday morning from a moly generator with 55.5 GBq (1.5 Ci) of Mo-99 yields approximately 47.2 GBq (1.28 Ci) of Tc-99m in 10 mL of normal saline (a common elution volume). By Friday morning of that same week, the same generator would be capable of delivering only about 17.2 GBq (0.47 Ci). The moly generator can be eluted more frequently than every 23 h; however, the Tc-99m yield will be less. Approximately half of the maximal yield will be available 6 h after the last elution. Figure 16-6 shows a typical time-activity curve for a moly generator.
1. Although the moly generator is by far the most widely used in nuclear medicine, other generator systems produce clinically useful radionuclides. When the half-life of the parent is much longer than that of the daughter (i.e., more than about 100 times longer), secular equilibrium occurs after approximately five to six half-lives of the daughter. In secular equilibrium, the activity of the parent and the daughter are the same if all of the parent atoms decay directly to the daughter.
2. Once secular equilibrium is achieved, the daughter will have an apparent half-life equal to that of the parent. The strontium-82/rubidium-82 (Sr-82/Rb-82) generator, with parent and daughter half-lives of 25.5 days and 75 s, respectively, reach secular equilibrium within approximately 7.5 min after elution. Figure 16-7 shows a time-activity curve demonstrating secular equilibrium (Table 16-2).
1. The users of moly generators are required to perform molybdenum and alumina breakthrough tests. Mo-99 contamination in the Tc-99m eluate is called molybdenum breakthrough. Mo-99 is an undesirable contaminant because its long half-life and highly energetic betas increase the radiation dose to the patient without providing any clinical information.
2. The high-energy γ-rays (approximately 740 and 780 keV) are very penetrating and cannot be efficiently detected by scintillation cameras. The U.S. Pharmacopeia (USP) and the U.S. Nuclear Regulatory Commission (NRC) limit the Mo-99 contamination to 0.15 µCi of Mo-99 per mCi of Tc-99m or (0.15 kBq/MBq) at the time of administration.
3. Contaminant limits are specified in 10CFR35.204 and include those for the Rb-82 generators: 0.02 µCi of Sr-85 per mCi of Rb-82 or (0.02 kBq/MBq).
 ▪ FIGURE 16-6 Time-activity curve of an Mo-99/Tc-99m radionuclide generator system demonstrating the ingrowth of Tc-99m and subsequent elution. The time to maximum Tc-99m activity, approximately 23 h, assumes there is no residual Tc-99m from a previous elution. Typical elution efficiency is approximately 85% (˜15% residual Tc-99m), and thus, time to maximum Tc-99m activity following the first elution is approximately 21 h. Approximately 50% of the maximum Tc-99m activity is obtained in 6 h. The maximum Tc-99m activity in the eluate is typically 80% to 90% of Mo-99 activity.  F. 16-9 F. 16-9 |
 ▪ FIGURE 16-7 Radionuclide activity as a function of time demonstrating the conditions of secular equilibrium.  F. 16-10 F. 16-10 |
TABLE 16-2 CLINICALLY USED RADIONUCLIDE GENERATOR SYSTEMS IN NUCLEAR MEDICINE | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
1. It is important to minimize radiation exposure to patients while preserving the diagnostic quality of the image. Radionuclides can be selected that have few particulate emissions and a high abundance of clinically useful photons.
2. Most modern scintillation cameras are optimized for photon energies close to 140 keV, which is a compromise among patient attenuation, spatial resolution, and detection efficiency. Photons whose energies are too low are largely attenuated by the body, increasing the patient dose without contributing to image formation. Highenergy photons are more likely to escape the body but have poor detection efficiency and easily penetrate collimator septa of scintillation cameras.
3. A radiopharmaceutical should have an effective half-life long enough to complete the study with an adequate concentration in the tissues of interest but short enough to minimize the patient dose.
1. The ability to detect and evaluate lesions depends largely on the concentration of the radiopharmaceutical in the organ, tissue, or lesion of interest or on a clinically useful uptake and clearance pattern. Maximizing the concentration of the radiopharmaceutical in the target tissues of interest while minimizing the uptake in surrounding (nontarget) tissues and organs improves contrast and the ability to detect subtle abnormalities in the radiopharmaceutical’s biodistribution.
2. Maximizing this target/nontarget ratio is characteristic of all clinically useful radiopharmaceuticals and is improved by observing the recommended interval between injection and imaging for the specific agent. This interval is a compromise between the uptake of the activity in the target tissue, washout of the activity in the background (nontarget) tissues, and practical considerations of clinic operations.
3. With some radiopharmaceuticals such as the bone scanning agent, Tc-99m-labeled methylene diphosphonate (99mTc-MDP), instructions to the patient, needed both to improve image quality and to reduce radiation dose to patient and technologist, might include the request to be well hydrated and to void the urinary bladder just prior to imaging.
4. Disassociation of the radionuclide from the radiopharmaceutical alters the desired biodistribution, thus degrading image quality. Good-quality control over radiopharmaceutical preparation helps to ensure that the radionuclide continues to be chemically bound to the pharmaceutical throughout the imaging session.
1. Low chemical toxicity is enhanced by the use of high-specific-activity, carrier-free radionuclides that also facilitate radiopharmaceutical preparation and minimize the required amount of the isotope.
2. For example, 3.7 GBq (100 mCi) of I-131 contains only 0.833 µg of iodine. Radionuclides should also have a chemical form, pH, concentration, and other characteristics that facilitate rapid complexing with the pharmaceutical under normal laboratory conditions.
3. The compounded radiopharmaceutical should be stable, with a shelf life compatible with clinical use, and should be readily available from several manufacturers to minimize cost.
1. Radiopharmaceuticals are also used for the treatment of a number of diseases. The goal of radiopharmaceutical therapy is to deliver a sufficiently large dose to the target organ, tissue, or cell type while limiting the dose to nontargeted tissues to minimize deterministic effects such as bone marrow suppression and to minimize the risk of cancer.
2. Prior to 2013, all approved therapeutic radiopharmaceuticals were based radionuclides that decayed either through beta-particle emission (32P, 89Sr, 90Y, 124I, 131I, 153Sm, 177Lu, 186Re, and 188Re) or Auger electron emission (103Pd, 111In, 117mSn, 123I, 125I, 193mPt, or 195mPt).
3. In May of 2013, the U.S. FDA approved the first therapy radiopharmaceutical using an alpha-particle emitter: Xofigo (radium Ra-223 dichloride) for the treatment of patients with castration-resistant prostate cancer (CRPC), symptomatic bone metastases, and no known visceral metastatic disease.
4. Since that milestone, there has been significant research activities and ongoing clinical trials in the development of other alpha-emitter therapy radiopharmaceuticals across a variety of cancer types such as breast, prostate, neuroendocrine tumors, and leukemia. Alpha emitters of current interest in radiopharmaceutical therapy include 211At, 212Pb, 212Bi, 213Bi, 223Ra, 225Ac, and 227Th.
|
1. Aside from the radionuclidic purity quality control performed on the 99mTc-pertechnetate generator eluate, the most common radiopharmaceutical quality control procedure is the test for radiochemical purity. The radiochemical purity assay identifies the fraction of the total radioactivity that is in the desired chemical form.
2. Radiochemical impurity can occur as the result of temperature changes, presence of unwanted oxidizing or reducing agents, pH changes, or radiation damage to the radiopharmaceutical (called autoradiolysis). The presence of radiochemical impurities compromises the diagnostic utility of the radiopharmaceutical by reducing uptake in the organ of interest and increasing background activity, thereby degrading image quality. In addition to lowering the diagnostic quality of the examination, radiochemical impurities unnecessarily increase patient dose.
3. The two principal radiochemical impurities in technetium-labeled radiopharmaceuticals are free (i.e., unbound) Tc-99m-pertechnetate and hydrolyzed Tc-99m. The Tc-99m radiopharmaceutical complex and its associated impurities will, depending upon the solvent, either remain at the origin or move with the solvent front to a location near the end of the strip.
1. Radiation doses to patients from diagnostic imaging procedures are an important issue and, in the absence of a medical physicist at an institution (e.g., private practice radiology), radiologists and nuclear medicine physicians are often consulted as the local experts. Radiation dosimetry is primarily of interest because radiation dose quantities serve as indices of the risk from diagnostic imaging procedures using ionizing radiation.
2. Dosimetry also plays an important role in radiopharmaceutical therapy where estimates of activity necessary to produce tumoricidal doses must be weighed against potential radiotoxicity to healthy tissue. In nuclear medicine procedures, the chemical form of the radiopharmaceutical, its route of administration (e.g., intravenous injection, ingestion, inhalation), the administered activity, the radionuclide, and patient-specific disease states and pharmacokinetics determine the patient dose.
1. The original mathematical methods, models, equations, nuclear decay data, and biokinetic parameters needed for computing radiation absorbed dose to internal organs were developed by the Medical Internal Radiation Dose (or MIRD) Committee of the Society of Nuclear Medicine and Molecular Imaging (SNMMI) in the late 1950s to early 1960s.
2. The most recent revision of the MIRD schema for internal organ dosimetry was published in 2009 as MIRD Pamphlet No. 21 in the Journal of Nuclear Medicine. In the 1970s, the International Commission on Radiological Protection (ICRP) developed its own set of expression, quantities, and terminology for the same purpose—estimating dose to internal organs following intake of both radionuclides and radiopharmaceuticals.
3. With the issue of ICRP Publication 130 (in 2015) and ICRP Publication 133 (in 2016), however, a harmonization of the dosimetry schema for internal organ dosimetry was made, thus providing a consistent terminology and approach to the field of internal dosimetry.
1. The mean absorbed dose D (rT) is defined as the mean energy imparted to target tissue (or region) rT per unit tissue mass. The rate at which the absorbed dose is delivered to target tissue rT within a patient from a radiopharmaceutical distributed uniformly within source tissue rS at time t following radiopharmaceutical administration is given as:

where A(rS, t) is the time-dependent activity of the radiopharmaceutical in source tissue rS and S(rT ← rS, t) is a quantity called the radionuclide S value (or S coefficient).
2. The expression involves a summation over all possible source regions rS that might contribute absorbed dose to the target region rT (Fig. 16-8).
3. For short-range radiations emitted by the radionuclide, the relevant source tissue may only be the target tissue itself (i.e., rT = rS).
4. For source tissues that emit photons (gamma-rays or x-rays), the relevant source tissues may be adjacent to or even at a distance from the target tissue (i.e., rT ≠ rS) and may also include the target tissue itself (i.e., rT = rS).
5. The time-dependent activity A(rS, t) represents the rate of nuclear decays in the source tissue rS at time t, while the S value represents the absorbed dose rate to the target tissue rT per radionuclide activity in source region tissue rS.
6. The S value is characteristic of the radionuclide and the anatomic model chosen to represent the internal body anatomy of the patient. In many cases, “reference” anatomic models are used to compute the S value. These may be anatomic models of either the averaged sized male or female patients at fixed ages—newborn, 1-, 5-, 10-, 15-year-old, and the adult (see Fig. 16-9).
 ▪ FIGURE 16-8 Illustration of source and target organ concept for calculation of the radiation absorbed dose to the lungs and thyroid gland (target organs) from a radiopharmaceutical (e.g., technetium-99m sulfur colloid) primarily located in the liver (source organ).  F. 16-11 F. 16-11 |
 ▪ FIGURE 16-9 Frontal views of the ICRP reference voxel phantom series as given in ICRP Publication 110 (adults) and Publication 143 (pediatric series).  F. 16-10 F. 16-10 |
1. The mean absorbed dose to a target tissue rT is thus obtained by time integrating the mean absorbed dose rate:


where Ã(rS, τ) is the time-integrated activity (TIA) in source tissue rS over dose integration period τ.
2. The time-integrated activity represents the total number of decays with the source tissue. The magnitude of the organ dose D(rT, τ) is directly proportional to the activity of the radiopharmaceutical administered to the patient, A0. Thus, doubling or tripling A0 will then double or triple the organ doses resulting from that nuclear medicine study.
3. It is then convenient to normalize organ dose by the value of A0, thus reporting the organ dose per activity administered (mGy per MBq). If the ratio of A(rS, t) to the administrated activity A0 is denoted as a(rS, t), then we can define the absorbed dose coefficient d(rT, τ) in the target tissue rT as:


where a(rS, t) is the fraction of administered activity in the source tissue rS at time t after administration, and the quantity ã(rS, τ) is the time-integrated activity coefficient (TIAC).
4. The time-dependent activity in source tissues of the patient may be obtained directly via quantitative imaging, including 2D planar imaging, 3D SPECT, 3D PET, or by tissue sampling (e.g., blood or urine collection). In the
case of preclinical animal studies, this may also be determined by the counting of individual organs or tissues of the experimental animals.
5. Alternatively, in the absence of direct measurement, the time-dependent activity in the source tissue may be obtained by numeric solution of a set of first-order coupled differential equations defined by compartment models for all organs and suborgan tissues of interest.
6. In earlier forms of the MIRD schema, the quantity ã(rS, τ) was referred to as the residence time of the radiopharmaceutical in the source tissue.
7. The second term in Equations 16-2 and 16-4 is the S value, a quantity in the MIRD schema that is specific to the radionuclide and to the computational anatomic model defining the spatial relationship and tissue compositions of rS and rT, as well as their intervening tissues. In equation form, the S value is defined as:

where Ei is the mean (or individual) energy per particle of the ith radiation emitted; Yi is number of the ith radiation emitted per nuclear decay; Δi is their product (mean energy of the ith radiation emitted per nuclear decay); φ(rT ← rS, Ei) is the absorbed fraction (fraction of Ei emitted in rS absorbed in target tissue rT); and m(rT) is the mass of the target tissue rT in the model.
8. The specific absorbed fraction Φ(rT ← rS, Ei) is further defined as the ratio of the absorbed fraction and the target mass (Table 16-3):


TABLE 16-3 Tc-99m S-VALUES (mGy/MBq-s) T. 16-5
T. 16-5
TARGET ORGANS
SOURCE ORGANS
Adrenals
Brain
LLI Contents
SI Content
Stomach Contents
ULI Contents
Heart Contents
Heart Wall
Kidneys
Liver
Lungs
Adrenals
1.80E-04
4.18E-10
2.25E-08
7.46E-08
2.73E-07
9.58E-08
2.53E-07
2.85E-07
7.24E-07
4.35E-07
2.33E-07
Brain
4.18E-10
4.23E-06
1.57E-11
3.91E-11
4.27E-10
4.68E-11
3.14E-09
2.54E-09
1.58E-10
8.16E-10
7.63E-09
Breasts
5.05E-08
3.17E-09
2.28E-09
7.35E-09
5.73E-08
8.00E-09
2.41E-07
2.61E-07
1.99E-08
6.82E-08
2.33E-07
Gallbladder wall
3.57E-07
1.54E-10
6.49E-08
4.38E-07
3.05E-07
7.53E-07
1.03E-07
1.22E-07
4.09E-07
8.70E-07
7.46E-08
LLI wall
1.98E-08
1.32E-11
1.23E-05
5.92E-07
9.10E-08
2.14E-07
4.06E-09
4.90E-09
5.50E-08
1.44E-08
3.29E-09
Small intestine
7.46E-08
3.91E-11
7.16E-07
4.22E-06
2.08E-07
1.25E-06
1.57E-08
2.06E-08
2.13E-07
1.16E-07
1.35E-08
Stomach wall
2.85E-07
2.52E-10
1.24E-07
2.13E-07
8.53E-06
2.86E-07
1.66E-07
2.65E-07
2.53E-07
1.48E-07
1.19E-07
ULI wall
9.41E-08
4.76E-11
3.10E-07
1.36E-06
2.65E-07
8.37E-06
2.12E-08
2.65E-08
2.12E-07
1.88E-07
1.81E-08
Heart wall
2.85E-07
2.54E-09
5.42E-09
2.06E-08
2.33E-07
2.97E-08
5.48E-06
1.19E-05
8.22E-08
2.33E-07
4.40E-07
Kidneys
7.24E-07
1.58E-10
7.10E-08
2.13E-07
2.73E-07
2.12E-07
6.45E-08
8.22E-08
1.32E-05
2.93E-07
6.66E-08
Liver
4.35E-07
8.16E-10
1.80E-08
1.16E-07
1.47E-07
1.87E-07
2.13E-07
2.33E-07
2.93E-07
3.16E–06
1.97E-07
Lungs
2.33E-07
7.63E-09
4.50E-09
1.35E-08
1.10E-07
1.77E-08
4.59E-07
4.40E-07
6.66E-08
2.09E-07
3.57E-06
Muscle
1.12E-07
2.21E-08
1.23E-07
1.12E-07
9.96E-08
1.07E-07
8.83E-08
9.20E-08
9.79E-08
7.52E-08
9.34E-08
Ovaries
3.14E-08
1.52E-11
1.26E-06
9.23E-07
5.85E-08
7.71E-07
4.55E-09
6.15E-09
7.02E-08
3.81E-08
5.39E-09
Pancreas
1.09E-06
4.15E-10
5.21E-08
1.42E-07
1.23E-06
1.62E-07
2.65E-07
3.57E-07
4.97E-07
3.86E-07
1.74E-07
Red marrow
2.53E-07
1.01E-07
2.01E-07
1.79E-07
7.50E-08
1.43E-07
1.11E-07
1.11E-07
1.71E-07
8.32E–08
1.11E-07
Osteogenic cells
2.67E-07
2.99E-07
1.82E-07
1.49E-07
1.03E-07
1.27E-07
1.60E-07
1.60E-07
1.62E-07
1.24E-07
1.66E-07
Skin
3.41E-08
3.97E-08
3.62E-08
3.01E-08
3.41E-08
3.09E-08
3.41E-08
3.70E-08
3.79E-08
3.62E-08
4.02E-08
Spleen
4.58E-07
5.19E-10
6.53E-08
1.01E-07
7.83E-07
1.05E-07
1.24E-07
1.67E-07
6.63E-07
7.22E-08
1.64E-07
Tests
1.54E-09
1.46E-12
1.40E-07
2.61E-08
2.90E-09
1.92E-08
5.16E-10
6.16E-10
3.10E-09
1.57E–09
3.67E-10
Thymus
5.66E-08
6.88E-09
2.04E-09
4.66E-09
3.65E-08
5.43E-09
8.87E-07
7.35E-07
1.73E-08
5.93E-08
2.85E-07
Thyroid
8.11E-09
1.35E-07
2.48E-10
4.87E-10
2.62E-09
7.69E-10
5.17E-08
4.33E-08
2.95E-09
8.64E-09
8.82E-08
Urinary bladder wall
7.55E-09
6.02E-12
4.98E-07
2.12E-07
1.73E-08
1.61E-07
2.22E-09
2.17E-09
1.87E-08
1.16E-08
1.33E-09
Uterus
1.89E-08
1.31E-11
5.17E-07
8.37E-07
5.05E-08
3.97E-07
4.87E-09
5.47E-09
6.42E-08
3.29E-08
4.10E-09
Total body
1.72E-07
1.25E-07
1.49E-07
1.59E-07
1.17E-07
1.41-07
1.17E-07
1.65E-07
1.58E-07
1.59E-07
1.44E-07
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree