Key Facts
Reflex Sympathetic Dystrophy
- •
Clinically, patients with reflex sympathetic dystrophy (RSD) present with intense, prolonged pain, vasomotor disturbances, delayed functional recovery, and trophic changes in the affected extremity that usually following minor trauma.
- •
Radiographs of RSD are characterized by regional osteoporosis with soft tissue swelling in the affected extremity. In the upper extremity, usually the hand and wrist, and in the lower extremity, the foot and ankle are most often involved.
- •
Radiographs can differentiate RSD from a primary articular disorder by the absence of significant intraarticular erosions and the preservation of the joint space in RSD.
- •
Three-phase bone scans are complementary to the clinical history in diagnosing RSD, characterized by increased accumulation of the radiotracer in the juxta-articular region of the affected joint on all three phases, with the delayed images being the most sensitive for confirming the diagnosis.
- •
Magnetic resonance imaging (MRI) adds very little to the clinical diagnosis of RSD, but it is beneficial in helping to exclude other entities that may present with similar clinical symptoms.
Regional Migratory Osteoporosis
- •
Clinically, patients with regional migratory osteoporosis (RMO) present with sudden onset of joint pain classically in the lower extremity and usually affecting men in the fourth to fifth decade.
- •
Diminishing pain in one joint with development of pain in an adjacent joint is typical of RMO.
- •
Osteoporosis of the affected area with preservation of joint space is typical of the radiographic findings of RMO or transient osteoporosis (TO).
- •
Classic findings on MRI are a bone marrow edema-like signal in the affected joint and a joint effusion. Findings return to normal following resolution of the patient’s symptoms.
- •
RMO must be differentiated from early osteonecrosis by MRI.
Osteogenesis Imperfecta
- •
Osteogenesis imperfecta is actually a spectrum of genetic disorders characterized by connective tissue defects. The Sillence type I is the most frequently observed variety and is characterized by blue sclera, ligamentous laxity, and increased tendency to sustain fractures.
- •
Osteogenesis imperfecta type II is the most severe form of the disease and is characterized by dwarfism and severe bone deformities. Few patients survive beyond the neonatal period.
- •
Osteogenesis imperfecta is the most common genetic cause for osteoporosis.
- •
Classically on radiographs, patients may have gracile bones, multiple fractures, and cystic changes at the metaphyses.
- •
Excessive callus at the healing fracture site may be distinguished from osteosarcoma with CT or MRI, which allows visualization of a fracture line confirming the diagnosis of fracture callus.
Osteoporosis is a very common disorder and the most frequently encountered metabolic bone disease worldwide. Diminished bone mass can result from a failure to reach optimal peak bone mass in early adulthood, increased bone resorption, or decreased bone formation after peak bone mass has been achieved. Numerous conditions can cause a decrease in bone mass; consequently, its distribution can aid in determining the underlying etiology. The distribution of osteoporosis can be divided into a regional and a generalized process. The regional form is confined to a segment of the body and associated with disorders of the appendicular skeleton. Classically, this is seen secondary to disuse or immobilization, but other causes include reflex sympathetic dystrophy and transient regional osteoporosis, which includes regional migratory osteoporosis. Numerous processes can result in generalized osteoporosis, which is primarily manifested in the axial skeleton and in the proximal portions of the appendicular skeleton. Common etiologies associated with the generalized form include senile causes, medications (e.g., corticosteroids and heparin), endocrine abnormalities, malnutrition/deficiency states, alcoholism, and liver disease, among others. The most common genetic cause of generalized osteoporosis is osteogenesis imperfecta. Through the combination of clinical history and laboratory information, as well as imaging findings, both the distribution and the underlying etiology for the development of osteoporosis can usually be determined, allowing for appropriate management and prevention of potentially serious complications.
REFLEX SYMPATHETIC DYSTROPHY
Reflex sympathetic dystrophy (RSD) is a pain syndrome that has various clinical forms, precipitating factors, physiopathologic hypotheses, and diagnostic criteria. It was first described in 1864 by Mitchell and his colleagues in U.S. Civil War soldiers who had suffered gunshot wounds that affected peripheral nerves. These patients went on to develop a persistent, burning pain with progressive trophic changes in the affected limb. More than six dozen different terms have been utilized since the original description to describe RSD in the English, French, and German literature. Some of these include causalgia, acute bony atrophy, Sudeck’s atrophy or osteodystrophy, post-traumatic osteoporosis, traumatic angiospasm or vasospasm, algodystrophy, reflex dystrophy of the extremities, minor causalgia, postinfarctional sclerodactyly, shoulder-hand syndrome, reflex neurovascular dystrophy, and the reflex sympathetic dystrophy syndrome . In 1994, a new term adopted by the International Association for the Study of Pain— complex regional pain syndromes (CRPS)—was intended to be an all-encompassing term for this group of disorders, in which pain that is felt to originate from the sympathetic nervous system is accepted as a common etiology. CRPS are subdivided into type I CRPS (RSD) and type II CRPS (causalgia). The distinction between the two is based on the absence of a documented nerve injury for causalgia. Because the older terminology is still widely used, the term RSD will be used herein.
Any neurally related condition that may affect the musculoskeletal system, including a neurologic or vascular insult, may serve as a potential source for developing RSD. Trauma secondary to accidental injury has been described as the most common cause of RSD. These injuries may include sprains, dislocations, fractures, traumatic amputations, crush injuries, contusions, and lacerations or punctures of the extremities. RSD will develop in 1% to 5% of these patients. Almost 28% of patients with a Colles fracture and 30% of patients with a tibial fracture will progress clinically to develop RSD. It is also observed in 1% to 20% of patients following myocardial infarctions and in 12% to 20% of patients following development of hemiplegia. Other reported causes may include infection, cervical osteoarthritis, tendinitis, peripheral neuropathy, herniated disks, use of barbiturates, and antituberculosis drugs, as well as psychological stress. Malignant tumors including those involving the brain, lung, ovary, breast, pancreas, and bladder are also associated with RSD. RSD has also been observed during the postoperative recovery period, commonly associated with arthroscopic surgical procedures and prolonged usage of extremity tourniquets.
RSD is observed less frequently in children than in adults. When present, it is more commonly found in girls than boys and will usually involve a lower extremity. It generally develops following a physical injury and may present with unique radionuclide features. The disease process is usually self-limited and benign in nature with no clinical residual.
The pathogenesis of RSD is not completely understood, and no pathophysiologic mechanism has been established. It is thought to be multifactorial in nature. The earliest and most widely accepted theory is that of the “internuncial pool” in which an injury or lesion will produce a painful impulse that travels via afferent pathways to the spinal cord, where a series of reflexes are initiated that spread via that interconnecting pool of neurons. These latter reflexes stimulate the lateral and anterior tracts, provoking efferent pathways that travel to the peripheral nerves producing the local findings of RSD. The International Association for the Study of Pain describes the CRPS as a complex neurologic disease involving the somatosensory, somatomotor, and autonomic nervous systems in various combinations, with distorted information processing of afferent sensory signals to the spinal cord. Others have proposed that the pain is caused by activation of sensory fibers by sympathetic efferents. The importance of the involvement of the sympathetic nervous system is further supported by the identification of nerve fibers in the periosteum that are sympathetic in origin and contain vasoactive intestinal peptide, a substance that stimulates bone resorption. Many other biochemical factors have also been proposed as being involved in this mechanism including substance P, histamine, and prostaglandins. Another theory is that minuscule peripheral nerve twigs are damaged in soft tissue injury and these twigs form artificial synapses; this is an attempt to explain why minor trauma may result in RSD.
Vascular abnormalities are well known to occur with RSD and are responsible for some of the changes seen on imaging exams. Many investigators feel that the sympathetic nervous system may also be responsible for these abnormalities; they confirmed their theories through the use of thermographic imaging that showed differences in the temperature of the affected extremities, which is felt to be related to blood flow. This was also confirmed by the significant effect on distal blood flow that occurs following sympathectomy. A different theory suggests that occlusion of the arterioles may initiate changes followed by capillary and venous stasis. This may occur following paralysis due to immobilization by casting or as described earlier, due to prolonged use of tourniquets in the operating room. This occlusion/stasis may result in local shunts that lead to the increased local temperature that is seen during certain phases of the disease. Despite all of these various proposals, the exact mechanism remains unknown.
The clinical signs and symptoms of RSD are variable ( Box 32-1 ). The diagnosis of florid RSD is generally not difficult, although the differential diagnosis is extensive. The diagnosis of a mild or early case is difficult due to the changing clinical features and the subjectivity of the clinical complaints.
- •
History of prior injury may be present
- •
Distal extremity affected
- •
Intense pain
- •
Vasomotor changes
- •
Delayed recovery
- •
Trophic changes
Early diagnosis of RSD is essential because the results of treatment are better when there is early initiation of therapy.
The most well-known form of RSD affects the upper extremity, with a regional distribution involving the distal forearm, wrist, and hand. It may also affect the lower extremity, most often at the foot and ankle. Classically, patients with RSD will present with four consistent clinical characteristics: intense prolonged pain, vasomotor disturbances, delayed functional recovery, and various trophic changes.
RSD is characterized by four clinical features: intense prolonged pain, vasomotor disturbances, delayed functional recovery, and trophic changes.
RSD is commonly divided into three stages. Not all patients will pass through all three stages. The initial (inflammatory) stage (I) lasts from 1 to 7 weeks and is characterized by diffuse nonfocal pain, inflammation, and edema associated with joint stiffness and decreased range of motion, as well as either hypothermia or hyperthermia. In the second (dystrophic) stage (II), which lasts from 3 to 6 months, the principal clinical findings are pain that decreases over time and is exacerbated with exercise, thickening of the skin and fascia, increased sensitivity of the skin to temperature and pressure changes, and the beginning of muscle atrophy. In the final (atrophic) stage (III), the pain persists and scleroderma-like skin changes occur with continued decrease in the range of motion and increased joint stiffness. Aponeurotic and tendinous retraction may occur in this final stage. The extremity becomes cooler with decreased vascularity. All of the changes in this final stage are irreversible. The final diagnosis of RSD is established by the patient’s clinical course and with the aid of the imaging findings ( Figure 32-1 ). The majority of cases completely resolve over time with few progressing to the final stage.


Imaging
Imaging of the patient with signs and symptoms of RSD is essential in helping to confirm the clinical assessment, as well as rule out potential serious entities that may present in a similar fashion ( Table 32-1 ). Imaging findings often reveal that RSD is a bilateral process, although the patient’s symptoms and imaging abnormalities will be much more pronounced on one side than the other. Rarely, a localized form may be observed affecting one joint in an extremity or one of several digits in a hand or foot.
| Radiographs | Soft Tissue Swelling |
| Trabecular resorption (spotty osteoporosis) | |
| Subperiosteal resorption (may mimic hyperparathyroidism) | |
| Endosteal resorption (scalloped appearance to the inner cortex) | |
| Cortical resorption (striated appearance to cortex) | |
| Subchondral resorption (mild erosive changes) | |
| Preservation of the cartilage space | |
| Usually no or minimal articular erosions | |
| Occasional insufficiency fractures | |
| Scintigraphy | All phases: Diffuse increased uptake with juxta-articular accentuation. |
| Unusual appearance in children. | |
| All three phases may show areas of decreased uptake (“cold”). | |
| MRI | Nonspecific. |
| Absent bone marrow edema in RSD, may aid in separating RSD from transient osteoporosis. | |
| May have joint effusion, skin changes, soft tissue edema, muscle atrophy, normal cartilage. |
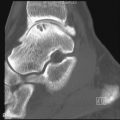
Along with the clinical findings, the radiographs may aid in making the diagnosis of RSD. The most common radiographic findings include soft tissue swelling and regional osteoporosis. The osteoporosis observed in association with RSD is nonspecific and is variably present; reported in 30% to 70% of RSD cases. The early findings of RSD are patchy demineralization of the epiphyses and short bones of the affected extremity. Genant and associates have described five types of bone resorption patterns that may occur with RSD, and these are best visualized with fine-detail radiography. These findings include irregular resorption of the trabecular bone in the metaphysis, resulting in patchy or spotty osteoporosis, subperiosteal bone resorption, intracortical bone resorption, endosteal bone resorption, and surface erosions of subchondral and juxtaarticular bone. The subperiosteal resorption is similar to that seen in hyperparathyroidism. Striation and tunneling of the cortical bone are observed with intracortical bone resorption, but these findings are not diagnostic of RSD and may occur with any condition that will result in disuse. The endosteal bone is the region of the greatest bone mineral loss in RSD and results in the initially observed excavation and scalloping at the endosteal surface. As a result, there is subsequent uniform remodeling of the endosteum and widening of the medullary canal. The erosions occurring in the subchondral and juxtaarticular bones result in small periarticular erosions and intraarticular gaps. Once patchy osteopenia is present, the patient has usually already progressed to stage II RSD.
A primary articular disorder may be mistaken for RSD due to the rapid onset and severity of bone resorption that may be observed in the periarticular regions. RSD can be differentiated from other arthritides due to the absence of significant intraarticular erosions and preservation of the joint space; the latter is a key finding in RSD. In rare cases, RSD may be accompanied by insufficiency fractures.
Bone Scintigraphy
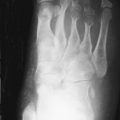
Bone scintigraphy is established as a beneficial imaging modality in making the diagnosis of RSD. Bone scintigraphy will demonstrate abnormalities that may be observed prior to recognizing abnormalities on radiographs ( Figure 32-2 ).




In patients with RSD, scintigraphic changes often precede radiographic abnormalities.
Imaging with Tc-99m MDP shows increased accumulation of the radiotracer in the involved bones. This is due to both increased vascularity, as well as rapid ion exchange at the highly vascularized bone surfaces. The classic distribution of the radiotracer is diffuse, with an accentuation of accumulation in the juxtaarticular regions of the involved extremity. This is preceded by hyperemia in a similar distribution, seen on both the immediate postinjection blood flow and the slightly delayed blood pool phases of the exam. With three-phase scanning, the delayed images show this pattern of periarticular accumulation, and this is the most sensitive indicator of the diagnosis ( Figure 32-3 ). Scintigraphic images obtained at different times may show migration of the abnormalities from one side of the joint to another similar to a pattern observed in regional migratory osteoporosis (RMO).




Rarely, decreased accumulation, or “cold” areas may be observed in bone scintigraphy examinations of children. The scintigraphic findings of this “cold” variant include photopenic abnormalities on the delayed scan and hypoxemia on the immediate blood-flow and blood-pool phases of the exam at the affected region. These abnormalities can be recognized in children who have open epiphyses by the incongruence of the involved epiphyseal activity compared with the remote ipsilateral or contralateral normal epiphyseal plate activity.
Magnetic Resonance Imaging
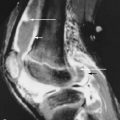
Very little data have been accumulated on MRI findings in patients with RSD. Koch et al. described nonspecific soft tissue changes such as edema and muscle atrophy in RSD patients and concluded that these findings were not helpful in establishing the diagnosis. Multiple other reports have described additional MRI findings that may aid in diagnosing RSD, including contrast enhancement of the involved joint/extremity and at a late stage, skin thinning ( Figure 32-4 ). Others have stated that an associated effusion in the involved joint is an additional useful sign in detecting RSD in its early stage. Bone marrow edema has been inconsistently seen in association with RSD. This is one of the features that may aid in distinguishing RSD from regional migratory osteoporosis, which typically shows bone marrow edema. The MRI examination is beneficial in confirming the clinical diagnosis of RSD, but it should not be used as the primary imaging modality in making this diagnosis.





MRI is helpful in excluding entities, such as fracture, that might be confused with reflex sympathetic dystrophy but is of limited use in establishing the diagnosis of RSD.
Differential Diagnosis
Clinically, the diagnosis of RSD may be difficult due to the variable clinical manifestations and the natural history of the disease. Many conditions may mimic the symptoms observed with RSD, including diabetic neuropathy and peripheral nerve damage, as well as cartilage injuries. The misdiagnosis of RSD as one of these other entities will lead to ineffective treatment in the early, more treatable stage, potentially resulting in permanent disability for the patient. Imaging studies may aid in making this diagnosis earlier and help confirm the clinical diagnosis. Based on the imaging findings, the differential diagnosis may include osteomyelitis, various arthritides, hyperparathyroidism, or even a trabecular fracture. Unfortunately, the usual findings observed on radiographs of decreased bone density with soft tissue swelling are nonspecific, and when observed may be a late manifestation of the disease. Bone scintigraphy adds to the diagnostic capability by allowing for earlier diagnoses to be made but is only 60% sensitive. MRI provides little beyond what may already be known clinically, but it does aid in narrowing the differential diagnosis by eliminating such entities as metastasis, bone infarcts, or even an occult fracture. Clinical follow-up remains the gold standard for the diagnosis of RSD with clinical confirmation based upon the patient’s response to therapy.
Treatment
The usual treatment consists of control of the patient’s symptoms, especially the pain, with nonsteroidal anti-inflammatory drugs. Veldman et al. suggested that early symptoms of RSD are responsive to this conservative treatment. Multiple other agents are being investigated to help in treatment, including vitamin C, clonidine, glucocorticoid therapy, and bisphosphonates, as well as ketamine and spinal cord stimulation. Ultimately, the diagnosis and treatment may rest on a nerve block using alpha-adrenergic blocking agents. Physical therapy is necessary to maintain full range of motion of the affected joints. Most patients will completely recover following conservative treatment with no persistent clinical sequela.
REGIONAL MIGRATORY OSTEOPOROSIS
Believed by some to be related to RSD, RMO is a sequential polyarticular arthralgia of the weight-bearing joints, most frequently seen in the lower extremities. It is an uncommon condition, although its incidence is probably underestimated, with many cases being labeled as transient osteoporosis of the hip. Along with transient osteoporosis of the hip, it is included in the conditions classified under transient regional osteoporosis. Transient regional osteoporosis is a general term applied to related conditions which are characterized by a sudden onset of joint pain followed by local osteopenia, which progresses to spontaneous healing.
The term transient regional osteoporosis includes the subtypes of transient osteoporosis of the hip and regional migratory osteoporosis. All of these conditions are characterized by sudden onset of joint pain followed by localized osteoporosis and eventual spontaneous healing.
RMO was first described by Duncan et al. in 1967 as a disorder affecting several joints at different times. They labeled this condition migratory osteolysis . It is characterized by a transient regional osteoporosis that is migratory in nature. The migratory nature of this process is the major factor that separates this entity from transient osteoporosis of the hip. RMO is an entity that is seen more frequently in men than women and is usually found during the fourth to fifth decade of life.
The etiology of this disease process is unknown and has been extensively debated. McCord et al. reported electromyographic (EMG) abnormalities in regional migratory osteoporosis, but EMG studies have been normal in other reports. Others have proposed that regional migratory osteoporosis and reflex sympathetic dystrophy (RSD) may be closely related or even identical diseases because RSD may precede the clinical onset of regional migratory osteoporosis. They have a similar pattern of osteoporosis early in the course. Radiographically, bilateral extremity joint involvement, as well as spread from one part of a joint to another can occur in both disorders. Similar scintigraphic findings are observed on bone scans in the two disorders. Clinical improvement may also occur with sympathetic blocks in both disorders.
A recently proposed hypothesis to explain the bone-marrow edema pattern observed on MRI is the regional accelerated phenomenon (RAP). In this proposed mechanism, areas of normal bone metabolism are rapidly increased to function between 2 and 10 times their normal rate as a response to noxious stimuli. The noxious stimuli may include microtrauma such as microscopic trabecular fractures due to stress or excessive loading of a mildly osteoporotic bone. A prolonged, exaggerated RAP can activate multiple different foci, where the normal bone repair mechanisms are most active, resulting in multifocal disease evidenced by the development of bone marrow edema along with the patient’s clinical symptoms. Although there are multiple proposed mechanisms for this disease, a definite consensus is still lacking.
Clinical Presentation
Clinically, patients with RMO have a moderate degree of tenderness over the affected joint, a joint effusion, and swelling with preservation of their full range of motion. Patients describe a history of acute or gradually increasing monoarticular joint pain with no related trauma or injury. The pain increases with weight bearing and reaches a maximum at approximately 2 months after the initial presentation. Weight-bearing-induced pain often results in a limp. In the ensuing weeks or months after initial presentation, additional joints may become involved. Often this will occur as the symptoms in the originally affected joint are resolving. The time interval varies between recurrences and has been reported as long as up to 2 years after the initial presentation. Usually the process progresses from adjacent joints, with the joint nearest the diseased one becoming the next to be affected. The clinically affected joints may overlap with more than one joint being affected at the same time, and rarely the same joint will be affected recurrently or with different parts of the joint being affected at different time intervals.
An isolated portion of a joint being affected at one time was first described by Lequesne et al., and was given the name partial transient osteoporosis . Two types of partial transient osteoporosis have been described, including the radial type where one or two rays of the hand or foot are involved, and the zonal type. In the zonal type, when the knee is involved, the affected portion of the joint will only be either the medial or lateral femoral condyle. Migration of the process to involve the entire joint or to involve another joint is observed. This migration in the zonal pattern resembles the classically described pattern of RMO. Classically described as a disease of the extremities, recent reports have described involvement of the axial skeleton, with associated spinal osteoporosis, which may be advanced and result in collapse of vertebral segments due to insufficiency fractures.
The clinical course can last up to nine months. Treatment consists of oral analgesics and non–weight-bearing strategies, as well as watchful waiting. A report has described a relapse of the disease following complete resolution of the initial episode.
Imaging
Radiographs
Radiographs will remain normal within the first several weeks ( Table 32-2 ). Four to eight weeks will be required to see visible changes, following the onset of symptoms, since osteopenia, which has begun with the initial presentation, is not yet severe enough until then for detection. Variable but often severe osteopenia will develop in the affected joint. The osteopenia progresses rapidly, subsequently diminishes, and then appears at other sites. In advanced cases, periarticular osteoporosis can be observed extending a large distance from the joint. Occasionally wavy periosteal bone formation can be seen along the shafts of tubular bones. The joint space is preserved which helps in differentiating this process from the advanced stages of avascular necrosis. Following the resolution of symptoms, bone remineralization occurs spontaneously after a 6-month to 8-month period.
| Radiographs | Osteopenia (after 4–8 weeks) |
| Cartilage space remains normal | |
| ± wavy periosteal reaction | |
| Scintigraphy | Increased activity on all three phases when disease is active. |
| Blood flow and blood pool images may show decreased activity with healing. | |
| MRI | Edema signal (low on T1W and high on T2W images) is seen in the bone marrow. |
| Joint effusion. | |
| No cartilage loss. |
Bone Scintigraphy
Initially in this disease process, there is increased activity in all three phases of the three-phase bone scan. This activity can be appreciated a few days after the onset of the patient’s symptoms, but before radiographic changes are evident. As the patient’s symptoms improve, the degree of hyperemia to the affected joint decreases, which is reflected as a relative decrease in the amount of activity on the perfusion and blood pool phases of the three-phase bone scan, when compared with the patient’s initial examination. There continues to be persistent uptake in the delayed phase of the examination due to the osteoblastic activity. This abnormality may be observed for months after the patient’s initial positive examination.
Magnetic Resonance Imaging
A diffuse bone marrow edema pattern can be observed with MRI, which can be detected as soon as 48 hours following the onset of clinical symptoms, similar to the three-phase bone scan. The changes are characterized by a homogeneous, ill defined area of mildly decreased signal on T1-weighted images and high signal on T2-weighted images ( Figure 32-5 ). These signal characteristics are thought to correspond to an increase in free water or edema within the fatty bone marrow. These marrow changes always abut the articular cartilage of the affected joint and extend into the metaphysis where the transition to normal marrow is ill-defined. In the femur, the marrow changes are generally seen affecting the head, neck, and intertrochanteric region. In the knee, the changes are observed more frequently in the lateral condyle ( Figure 32-6 ). The tarsal bones are commonly affected, but the metatarsal bones will rarely be involved. A small joint effusion may be observed in the affected joints. Following symptomatic and supportive therapy a regression of the MRI signal abnormalities can be seen without the persistence of identifiable abnormalities within the bone marrow.