Chapter 18 Renal Tumors
Introduction*
Cancer of the kidneys constitutes nearly 2% of the total human cancer burden occurring in all world regions, with a slight increased incidence in developed countries. Renal neoplasms represent several histologic subtypes that show a characteristic pattern of somatic mutations, which along with histopathology, constitutes the major criteria for their classification. The 2004 World Health Organization (WHO) classification of adult renal neoplasms1 is based on pathology and genetic abnormalities and recognizes several newer subtypes. In addition, renal tumors are a feature in several inherited cancer syndromes including von Hippel–Landau (vHL) disease and Birt-Hogg-Dubé syndrome.
Epidemiology and Risk Factors
RCC constitutes over 90% of all malignancies occurring in the kidney in adults. Overall, it is the 12th most common site in men and 17th in women and represents 2% of all solid cancers in adults worldwide.2 In males living in industrialized countries including Japan, it is more common, ranking 6th along with non-Hodgkin’s lymphoma, whereas in less developed countries, it is 16th. In the United States, it is the 7th most common site in males and the 8th most common site of cancer in females with 13,120 deaths and 60,920 new cases expected in the United States in 2011.3
The incidence of RCCs has been gradually increasing each year in both the United States and Europe over the past few decades. This is not entirely accounted for by increased detection of incidental carcinomas owing to frequent use of imaging studies because the incidence of advanced RCC has also increased over time.4 RCC is nearly twice as common in men as in women and in whites as in blacks. However, over the past few decades, there has been a rapid increase in incidence in women and also in blacks, thereby tilting these ratios. Age-adjusted incidence rates of RCC among white men, white women, black men, and black women in the United States during the period 1992 to 2002 were 13.8, 6.6, 16.8, and 8.0 per 100,000 person years, respectively.3,4
Rates of RCC vary considerably internationally, sometimes by up to a factor of 10, with increased incidence in several Western countries and Australia/New Zealand and the lowest rates in Asia and Africa. These demographic trends highlight the significant role of exogenous risk factors.2,5
Several exogenous and endogenous factors appear to contribute to the development of RCC (Table 18-1). Cigarette smoking is an established causal risk factor for developing RCC and is estimated to account for nearly 20% to 30% of cancers in men and 10% to 20% cancers in women. A meta-analysis including more than 20,000 patients estimated the relative risks of 2.0 and 1.6 among heavy smokers for males and females, respectively, with a slow decline in risk with several years of cessation.6
Table 18-1 Risk Factors for Developing Sporadic Renal Cell Carcinoma
| Cigarette smoking |
| Obesity |
| Dietary factors |
| Hypertension and antihypertensive medications |
| Analgesics |
| Hormonal and reproductive factors |
| Kidney transplantation and dialysis |
| Radiation |
Several studies have now established compelling evidence that links obesity to an excess risk of RCC. It is estimated that the relative risk of RCC is increased by 1.07 per unit increase in body mass index.7 Obesity is considered to be a causal factor in nearly 30% to 40% of all RCCs in the Western world.4 The mechanism underlying the pathways of association between obesity and RCC is not clear, but several metabolic and hormonal pathways such as increased levels of peptide and steroid hormones, estrogen, or elevated levels of insulin-like growth factor-1 (IGF-1) and interleukin-6 (IL-6) pathway have been hypothesized as possible associations.
The differences in incidence of RCC between Asia and the West suggest that diet may have an important role in the etiology of RCCs. In general, diets low in fat and rich in fruits and vegetables have a protective effect.8 There is an increased incidence of RCC in patients with hypertension and the odds ratio is calculated by many authors to be 1.3 to 2.0. It is now thought that hypertension-induced renal injury is the risk factor, although the exact mechanism is unknown.4,8 Although historically, analgesic use, especially phenacetin, was linked to the risk of RCC, this is not proved. Ionizing radiation is weakly linked to the development of RCC and this increased incidence has been recorded in patients who received treatment for ankylosing spondylitis and cervical cancer.4
Anatomy
The kidneys are paired structures situated in the retroperitoneum on either side of the vertebral bodies. Whereas most individuals have single renal arteries, multiple renal arteries are the most common anatomic variant, seen in up to 30% of normal individuals.9,10
Bilateral multiple renal arteries occur in approximately 10% to 15% of the population. Accessory renal arteries most commonly arise from the abdominal aorta and are classified as polar (supplying one of the poles) and hilar (running along with the main renal artery). The polar accessory renal arteries are usually slender, but the hilar renal artery is not always smaller than the main renal artery. Rarely, the accessory renal arteries arise from the celiac, mesenteric, lumbar, middle colic, or middle sacral artery.11
The left renal vein receives the gonadal and lumbar veins before draining into the inferior vena cava (IVC) anterior to the aorta and posterior to the superior mesenteric artery. The right renal vein, usually shorter than the left, directly drains into the IVC. Renal veins usually lie anterior to the arteries at the hilum. Multiple renal veins are the most common anatomic variant, seen in 15% to 30% of the population and are more common on the right side. The most common anomaly of the left renal vein is the circumaortic renal vein, seen in 2% to 17% of the population, in which the left renal vein divides into ventral and dorsal limbs that encircle the aorta. A rarer anatomic variant, the retroaortic renal vein, is seen in 2% to 3% of the population.11
Pathology
It is now well understood that RCC is actually a family of related tumors with different histologies, prognoses, and therapies. The 2004 WHO classification describes various categories and entities based on pathologic and genetic analyses and builds upon the previous classifications including the previous Heidelberg classification. Histologic subtypes include clear cell (“conventional”) adenocarcinoma (80%), papillary (15%), chromophobe (5%), collecting duct (1%), and unclassified (4%).1,12,13 Major changes in the 2004 WHO classification include emphasis placed on defining familial RCC, carcinoma associated with Xp11 translocations, carcinoma associated with neuroblastoma, multilocular cystic RCC, tubular, mucinous and spindle cell carcinoma, and mixed epithelial and stromal tumor (Table 18-2).
Table 18-2 World Health Organization Classification of Renal Tumors
| Clear cell carcinoma |
| Multilocular clear cell RCC |
| Papillary RCC |
| Chromophobe RCC |
| Renal medullary carcinoma |
| Xp11 translocation carcinoma |
| Carcinoma associated with neuroblastoma |
| Mucinous tubular and spindle cell carcinoma |
| RCC unclassified |
RCC, renal cell carcinoma.
From Eble JM, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics of tumors of the urinary system and male genital organs. World Health Organization Classification of Tumors. Lyon, France: IARC Press; 2004.
Clear cell carcinoma is the most common subtype, accounting for nearly 80% of all RCCs. Papillary RCC (15%) and chromophobe (5%) are the second and third most common subtypes. Each of these subtypes has differing cytogenetic and immunohistochemical profiles and each is thought to arise from a different part of the nephron. Histopathologic grading of the nuclei of the tumor is made by dividing them into the four-tier Fuhrman nuclear classification14 with grade I being the best-differentiated and grade IV the most anaplastic. There is a separate category of spindle-shaped cells with ominous prognostic significance seen in all histologic subtypes, referred to as a sarcomatoid variant. Clear cell carcinoma displays large, uniform cells with abundant clear cytoplasm rich in glycogen and lipid (Figure 18-1). All kidney tumors of clear cell type of any size are considered malignant. Most clear cell carcinoma are solitary cortical neoplasms. They may be multicentric in up to 4% and bilateral in 0.5% to 3%. The term granular cell carcinoma, indicating acidophilic cytoplasm, was an entity in the 1998 WHO classification and is now considered to be clear cell carcinoma based on clinical and pathologic similarities. Clear cell carcinoma is typically highly vascular and has the worst overall prognosis. Defects in the vHL suppressor gene are reported in over 60% of sporadic cases of clear cell carcinoma.15 The vHL gene is found on the short arm of chromosome 3, at 3p25. vHL protein, the product of this gene, is a potent tumor suppressor. Its loss, mutation, or inactivation in turn leads to higher levels of a transcription factor named hypoxia-inducible factor (HIF), which is produced constitutively but bound to and degraded by functional vHL in normoxic conditions. In hypoxic conditions, or in absence of functional vHL, high levels of HIF will result in the transcription and translation of several proteins involved in tumor angiogenesis, including vascular endothelial growth factor (VGEF).
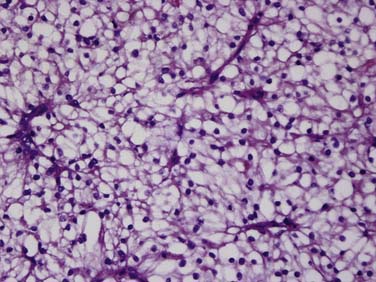
Figure 18-1 Clear cell carcinoma. Histology image (×100 magnification) shows abundant clear cells filled with glycogen.
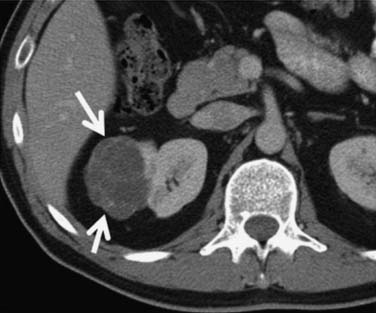
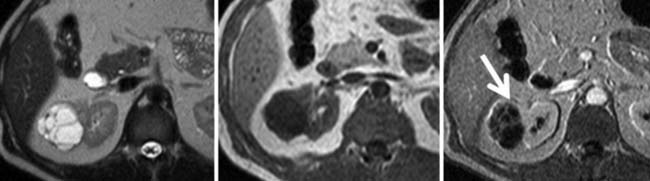
Multilocular cystic RCC is a variant of conventional clear cell tumors characterized by a cystic mass with less than 25% solid components and entirely composed of cysts of variable size separated from the kidney by a fibrous capsule (Figures 18-2 and 18-3).16,17 These tumors tend to be indolent and have a very low metastatic potential.
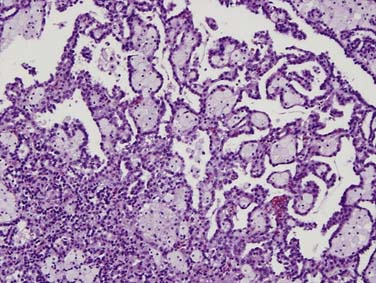
Papillary tumors are characterized by abnormalities of chromosomes 7 and 17 and loss of chromosome Y. They typically have a papillary growth pattern and exhibit a frondlike appearance on histologic examination (Figure 18-4). There is a 5:1 male predominance. Hereditary papillary RCC is an autosomal dominant form of the disease associated with multifocal papillary RCC.18 Papillary RCCs may have a less aggressive course than the clear cell RCC and are subdivided into cellular type 1 and type II. These tumors have cells arranged over a fibrovascular core and a papillary growth pattern. These terminologies are preferred over the terms “basophilic” (type 1 papillary) and “eosinophilic” (type 2 papillary) RCC used in previous classifications. Type 2 papillary RCC is typically of a higher grade and more aggressive than type 1. Once metastatic, patients with papillary RCC fare more poorly than patients with clear cell histologies, owing to the absence of effective systemic therapies.
Chromophobe RCCs (Figure 18-5) are a less aggressive subtype with 86% of tumors being T1 or T2 at diagnosis. The relationship between oncocytoma and chromophobe RCC is still under investigation and both are considered to be derived from the intercalated cells of the collecting duct. The Birt-Hogg-Dubé syndrome is a form of inherited RCC also characterized by hair follicle hamartomas. Patients with this syndrome often have chromophobe tumors or mixed chromophobe tumors and oncocytomas.19
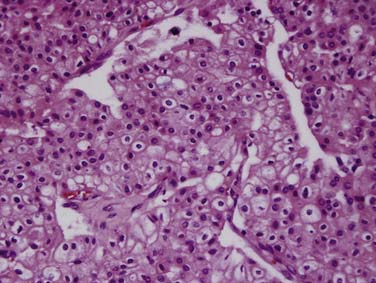
Figure 18-5 Chromophobe RCC. Histology specimen shows prominent pale cytoplasm with perinuclear halos.
Sporadic oncocytomas are relatively common and are considered benign but indistinguishable from RCCs on imaging (Figure 18-6).
Collecting duct tumors, which arise from the medullary collecting duct, often occur in younger patients and frequently present with advanced disease at the time of diagnosis. Renal medullary carcinoma is a rare subtype, closely related to collecting duct carcinoma, with a very poor prognosis, that occurs in young patients with sickle cell trait.20,21
RCC has a propensity for extending, as tumor thrombus, into the tributaries of the renal veins and subsequently to the main renal vein, IVC, hepatic veins, and potentially the right atrium. Rarely, the tumor may extend directly into the ipsilateral adrenal gland, or adjacent musculature, liver, spleen, pancreas, and colon. Hematogenous metastases are more common and occur earlier than lymphatic dissemination, the former most commonly to the lungs and bone, but essentially to any organ, including the subcutaneous tissues and skeletal muscle. Lymphatic dissemination, however, can involve regional nodes and spread to the chest via the cisterna chylii with mediastinal nodal involvement.
Staging
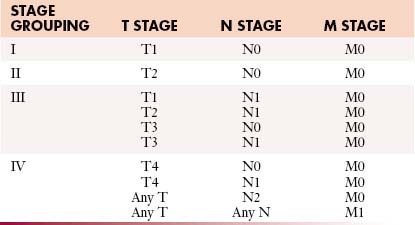
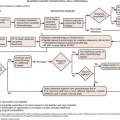
Staging systems are designed to reflect the modes of spread (Figure 18-7) and are used to stratify treatment options and prognoses and assess survival characteristics. The current version, the 2009 tumor-node-metastasis (TNM) staging of the American Joint Committee on Cancer (AJCC) and the International Union Against Cancer (UICC) is presented in Tables 18-3 and 18-4 and Figure 18-7.3,22
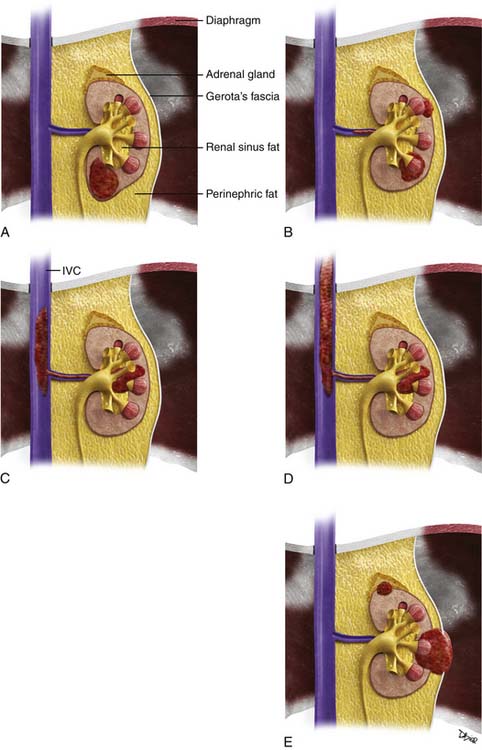
Figure 18-7 A-E, T staging according to the American Joint Committee on Cancer tumor-node-metastasis (TNM) staging system for RCC. Tumors confined to the kidney are staged as T1 when smaller than 7 cm and as T2 when larger than 7 cm. Tumors invading the renal sinus fat or perinephric fat are staged as T3a, and tumors involving Gerota’s fascia and adrenal gland are staged T4. IVC, inferior vena cava. See Table 18-3.
Table 18-3 Tumor-Node-Metastasis Staging for Renal Cell Carcinoma
| STAGE | CHARACTERISTICS |
|---|---|
| Primary Tumor (T) | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor confined to the kidney < 7 cm in greatest dimension |
| T1a | Tumor confined to kidney < 4 cm in greatest dimension |
| T1b | Tumor confined to kidney > 4 cm but < 7 cm in greatest dimension |
| T2 | Tumor confined to kidney and > 7 cm in greatest dimension |
| T2a | Tumor confined to kidney > 7 cm but < 10 cm in greatest dimension |
| T2b | Tumor confined to kidney > 10 cm |
| T3 | Tumor extends into major veins or perinephric tissues but not into the ipsilateral adrenal gland and not beyond Gerota’s fascia |
| T3a | Tumor grossly extends into the renal vein or its segmental (muscle containing) branches, or invades perirenal and/or renal sinus fat but not beyond Gerota’s fascia |
| T3b | Tumor grossly extends into the vena cava below the diaphragm |
| T3c | Tumor grossly extends into the vena cava above the diaphragm or invades the wall of the vena cava |
| T4 | Tumor invades beyond Gerota’s fascia (including contiguous extension into the ipsilateral adrenal gland) |
| Regional Lymph Nodes (N) | |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Metastasis in regional lymph node(s) |
| Distant Metastasis (M) | |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
From Edge SB, Byrd DR, Compton CC, et al, eds. AJCC Cancer Staging Manual. 7th ed. New York: Springer; 2010.
Prognosis is generally reflected in staging severity (Table 18-5) with lower stage disease associated with longer survival rates. For example, a patient with a T1a N0 M0 lesion has a 90% to 100% 5-year survival, whereas the survival for a patient with a T3b-c N0 M0 lesion decreases to 40% to 65%. Systemic metastases at the time of presentation are associated with only a 0% to 20% 5-year survival. The histologic type of tumor is an important consideration not included in the TNM system. For example, the time from nephrectomy to metastasis is twice as long for patients with chromophobe tumors than for those with clear cell or papillary subtypes.23
Table 18-5 Survival by Tumor-Node-Metastasis Stage
| DISEASE EXTENT | TNM STAGE | 5-YEAR SURVIVAL (%) |
|---|---|---|
| All organs confined | T1-T2 N0 M0 | 70-90 |
| ≤4 cm | T1a N0 M0 | 90-100 |
| 4-7 cm | T1b N0 M0 | 80-90 |
| ≥7 cm | T2 N0 M0 | 70-80 |
| Invasion of perinephric fat | T3a N0 M0 | 60-80 |
| Adrenal involvement | T3a N0 M0 | 0-30 |
| Venous involvement | T3b-c N0 M0 | 40-65 |
| Locally advanced | T4 N0 M0 | 0-20 |
| Lymphatic involvement | Any T, N+, M0 | 0-20 |
| Systemic metastases | Any T, Any N, M1 | 0-10 |
Imaging Evaluation
It is generally considered that a risk-adapted approach is used for workup of possible sites of metastases. Chest CT should be performed if the primary tumor is large or locally aggressive, because metastases are more common in these patients.24 Plain chest radiography without CT should be reserved for patients with a very low risk of metastatic disease or for those in long-term follow-up. Brain MRIs and nuclear medicine bone scans are generally only justified if there are symptoms and signs to suggest disease at these sites or if the tumor is large and locally aggressive (although even the latter is debated).25 Technetium-99m methylene diphosphate (99mTc-MDP) bone scans, however, may have limitations in detecting the typical osteolytic bone metastases from RCC. The value of pelvic CT in staging is limited.26
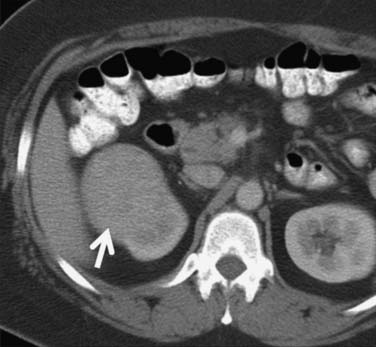
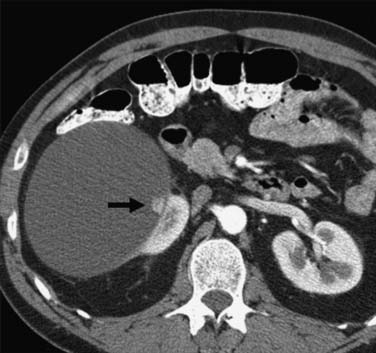
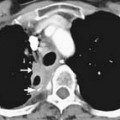
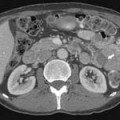
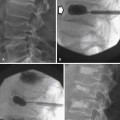
Calcification, which can be detected on an abdominal radiograph in as many as 20% of renal adenocarcinomas, is even easier to detect on an unenhanced CT examination (Figure 18-8). However, calcification also may be seen in other renal masses, including common lesions such as benign cysts. In all but very large tumors, renal function is preserved. In fact, diminished renal function suggests tumor thrombus in the renal vein. The tumor is identified as a mass with mottled density that causes a focal bulge in the renal contour. Such tumors are termed exophytic (Figure 18-9). Tumors that do not deform the renal contour are termed intrarenal or central (Figure 18-10). Tumors that have both an exophytic and a central component are referred to as mixed. Involvement of the collecting system is seen as irregularity of the urothelial surface or complete occlusion of a calyx or infundibulum. A filling defect within the collecting system may represent a blood clot or tumor invasion but is relatively uncommon with renal parenchymal tumors. In fact, if a mass is present in the collecting system, the lesion is more likely to represent a transitional cell carcinoma that has invaded the renal parenchyma rather than an RCC that invades the collecting system. Some tumors, particularly those with sarcomatoid features, have an infiltrative appearance in which the reniform shape of the kidney is preserved (Figure 18-11)
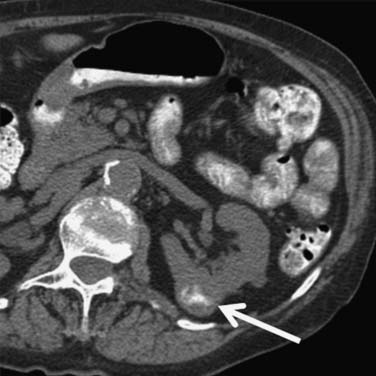
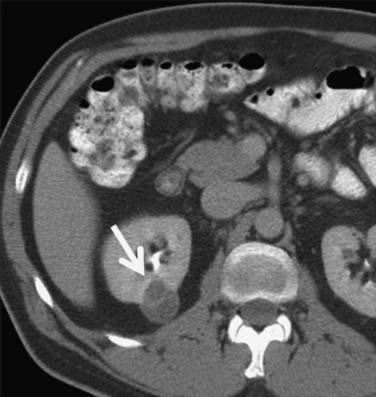
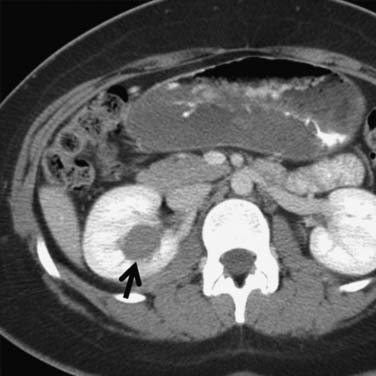
Figure 18-10 Axial CT scan of the abdomen with IV contrast shows a central tumor (arrow) in the right kidney.
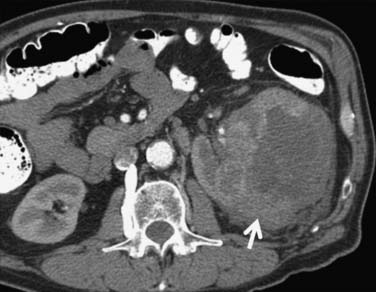
Occasionally, the tumor can be found arising in the wall of a simple renal cyst (Figure 18-12). In some cases, the tumor itself has a cystic appearance and may be difficult to distinguish from a complex but benign renal cyst. Hartman27 estimates that 15% of cases of RCC are cystic on radiologic and pathologic examination. The clinical features of cystic RCC are similar to those that are completely solid. The majority will have clear cell histology, but some may also be papillary.
Computed Tomography Evaluation
Suggested protocols for preoperative evaluation and follow-up, using multislice computed tomography (MSCT) are discussed. In addition to assessing tumor staging, preoperative evaluation focuses on delineating the tumor with particular attention to the relationship of the tumor to adjacent structures, including vascular relationships. In comparison, follow-up evaluation is directed toward surveillance for residual or recurrent disease. In general, 100 to 150 mL of iodinated intravenous (IV) contrast medium is used, with a flow rate of 2 to 3 mL/sec.
Preoperative Computed Tomography Evaluation
During the nephrographic phase, the contrast, filtered through the glomeruli, enters the loop of Henle and the collecting ducts. It is to be noted that the nephrographic phase can occur earlier in patients injected with contrast at a rapid rate. During the nephrographic phase, renal medullary enhancement is similar to that of the cortex. The excretory phase begins when the contrast is excreted into the calyces at approximately 3 to 5 minutes. Although the nephrograms remain homogeneous in the excretory phase, the attenuation decreases uniformly owing to a decrease in the plasma concentration of contrast and continued excretion from the kidneys.28 The corticomedullary phase and all the subsequent phases can last longer in patients with renal dysfunction and in those with diminished cardiac output.
Multiplanar re-formatted and three-dimensional (3D) volume-rendered presentations of the renal phase images are helpful in allowing appreciation of the relationships of structures, particularly for surgeons (Figure 18-13). Such re-formations are best obtained with the thinnest possible collimation and some degree of reconstruction interval overlap (typically, <1.5 mm interval and 10-50% overlap), and transferred to a workstation for the purpose of creating multiplanar re-formatted images, 3D volume rendering, and maximum intensity projection (MIP) images. The late-arterial phase provides a useful angiographic image of arterial and venous supply to the kidney, but it has limited additional utility for lesion detection and characterization when a nephrographic phase is employed. The combination of excretory and nephrographic phases have been shown to improve lesion detection and staging.29,30
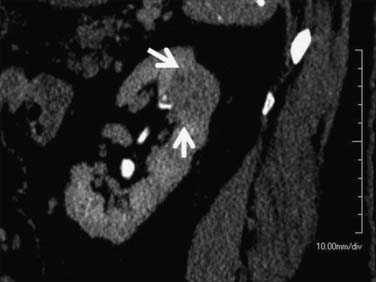
It has been shown by several investigators that clear cell RCC enhances to a greater extent and is more heterogeneous in appearance than other histologic subtypes of RCC (Figure 18-14). Kim and coworkers31 showed that an increase in attenuation of 84 Hounsfield units in the corticomedullary phase differentiated clear cell RCC from non–clear cell tumors with a sensitivity of 74% and a specificity of 100%. Herts and colleagues32 showed that papillary tumors were more homogeneous and had a much lower tumor-to-parenchyma enhancement ratio than was present in nonpapillary subtypes, particularly with tumors less than 3 cm in diameter (Figure 18-15). Chromophobe tumors also are less hypervascular than clear cell tumors and tend to have a more peripheral pattern of enhancement; their appearance, however, is not sufficiently characteristic to allow them to be reliably differentiated from papillary lesions (Figure 18-16). Oncocytomas cannot be reliably differentiated from RCCs by imaging and, thus, are also considered to be surgical lesions (see Figure 18-15). The well-known prominent central scar that can be used to suggest the diagnosis may also be present in necrotic clear cell tumors. Collecting duct and medullary RCC tumors are located centrally within the kidney and show variable, typically limited, postcontrast enhancement (Figures 18-17 and 18-18
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


