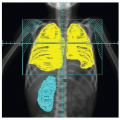
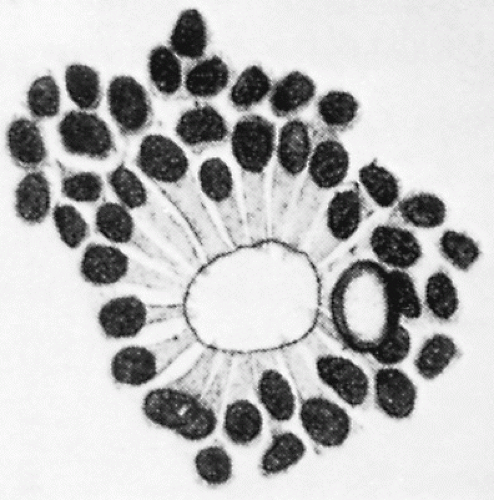
Retinoblastoma (RB) is the most common malignant intraocular tumor of childhood. The tumor is of neuroepithelial origin and arises from the nucleated layers of one or both eyes (1). RB consists of undifferentiated small anaplastic cells, which may be round or polygonal. Scant cytoplasm surrounds the large nuclei, which characteristically stain deeply with hematoxylin. Calcification commonly occurs in necrotic areas (2). Both Flexner and Wintersteiner described the arrangement of the more differentiated malignant cells of RB in neuroepithelial rosettes. These Flexner-Wintersteiner rosettes appear to represent an attempt to differentiate into photoreceptor cells (Fig. 5.1) (3).
As RB grows, it may cause a retinal detachment secondary to a solid or multifocal mass (endophytic type of growth). Endophytic tumors may break through the inner layers of the retina to the vitreous. The tumor may also form a pedunculated mass (exophytic type of growth). Both patterns of growth may occur in the same eye, and neither one is of prognostic significance (4). It is not unusual for RB to seed the vitreous. These vitreous seeds may grow even though they lack a blood supply (2).
Figure 5.1 A rosette characteristic of differentiated retinoblastoma, after a drawing by Wintersteiner. Note the cylindrical nuclei and fine protoplasm extensions toward the lumen. (From Dudgeon J. Retinoblastoma: trends in conservative management. Br J Ophthalmol. 1995;79:104, with permission.)
INCIDENCE
The reported incidence of RB ranges from 1 in 14,000 live births to 1 in 34,000 (3,5). There are approximately 200-350 new cases per year in the United States and 50 in the United Kingdom (2,3, 4, 5, 6, 7). The tumor may be more common in Latin America (8).
RB shows no predilection for sex, race, or the right or left eye. Between 65% and 80% of cases are unilateral (2,3,9, 10, 11, 12). The frequency of bilateral tumors is higher at institutions serving as referral centers for more complicated cases. Bilaterality may be ascertained concurrently or sequentially.
The diagnosis may be made from shortly after birth until 7 years of age. The mean age of detection is 2-4 months, and most cases are discovered before 3 years of age (2,3,13,14). In general, unilateral RB is diagnosed at a later age than is bilateral disease. In the Mayo Clinic series the median ages at diagnosis were 4.5 months for bilateral disease and 22 months for unilateral disease, an observation supported by others (4,7,15).
THE GENETICS AND MOLECULAR BIOLOGY OF RETINOBLASTOMA
The infectious nature of some cancers was demonstrated by Francis Peyton Rous (1879-1970). In a 1910 experiment he showed that submicroscopic, filterable agents (viruses) isolated from a chicken sarcoma could induce new sarcomas in healthy chickens. Rous’s work languished in obscurity before it was rediscovered and recognized with the Nobel Prize in Physiology or Medicine in 1966. In his Nobel lecture, “The Challenge to Man of the Neoplastic Cell,” Rous (16) considered the possible existence of growth-promoting genes. He called them oncogens; today they are called oncogenes.
Rous was wrong about oncogenes. Theodor Boveri (17), on the other hand, was right. In 1914, he used studies of normal mitosis in sea urchins and worms as a platform for suggesting that cancer might be caused by the abnormal gain or loss of chromosomes and their function.
Boveri predicted that the genetic abnormalities leading to the development of cancer are of two sorts: growth-promoting genes and growth-suppressing genes. If the growth-promoting genes are excessive in number or activity, they lead to cell proliferation. If the growth-suppressing genes are defective in amount or activity, they fail to halt cell proliferation and lead to unbridled cell replication. These growth-promoting genes are called oncogenes. The growth-suppressing genes are called tumor suppressor genes.
We may think of oncogenes and tumor suppressor genes as analogous to the accelerator and brake pedals of an automobile. When it is idling with the transmission in drive, one can move the car forward by pushing on the accelerator pedal, by taking pressure off the brake pedal, or by doing both simultaneously. Similarly, cell growth and proliferation, leading to cancer, can occur by the activity of the oncogenes or the inactivity of the suppressor genes (17, 18, 19, 20). Uncontrolled cell growth, or cancer, may be thought of as a set of physiologic controls of cell growth gone awry.
In 1972, Alfred G. Knudson Jr proposed a simple genetic model to explain the origins of RB. Almost 30 years after the original article was published, Knudson described the origins of his discovery.
I was interested in the fact that the germ-line mutation, which is a de novo mutation in 80% of the germ-line mutants, is not a sufficient condition for tumorigenesis— some children with an affected parent do not develop a tumor, but later produce an affected child, indicating that they carried the germ-line mutation. Most affected children with an affected parent develop tumors bilaterally but some do so unilaterally. Approximately 60% of all cases are unilateral in the United States and do not carry a predisposing germ-line mutation. I calculated that the numbers of tumors per heritable case fall in a Poisson distribution with a mean of 3. F rom this, it can be inferred that 5% (e3 = 0.05) of carriers of the germ-line mutation developed no tumor, which fits approximately with observation. The distribution of bilateral cases that have not yet been diagnosed (S) at different ages showed a linear decline on a semi-log plot. That is, ln S = kt (where k is a constant that incorporates the mutation rate and t is time), as expected for a one-hit phenomenon. From this, I predicted that hereditary retinoblastoma involves two mutations and, knowing that one of these had to be a germ-line mutation, I hypothesized that the other one would be somatic. The unilateral cases with no positive family history, only a minority of whom carries a germ-line mutation, showed a distribution consistent with two mutations, so both of these ought to be somatic. The hereditary and nonhereditary forms of the tumor seem to entail the same number of events—a hypothesis that became known as the “two-hit hypothesis” (21).
Knudson (18,19) sought to understand how the disease might have a familial (i.e., hereditary) form and a sporadic (i.e., nonhereditary) form. In a retrospective statistical analysis, he plotted the logarithm of the proportion of cases not yet diagnosed against the age for bilateral (i.e., hereditary) and unilateral (largely nonhereditary) forms of RB. The graph for the bilateral type generated a straight line, whereas the unilateral type created a curved line with second-order kinetics. Knudson inferred that the bilateral type was explicable by a single random somatic event acting in the presence of an existing germ-line mutation, whereas the unilateral form resulted from two somatic events. He also analyzed the number of tumors in patients with bilateral RB. He found that the mean number was 3 and that the distribution of the number of tumors followed the Poisson equation. He concluded that the tumor events were random and independent (18,19).
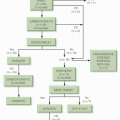
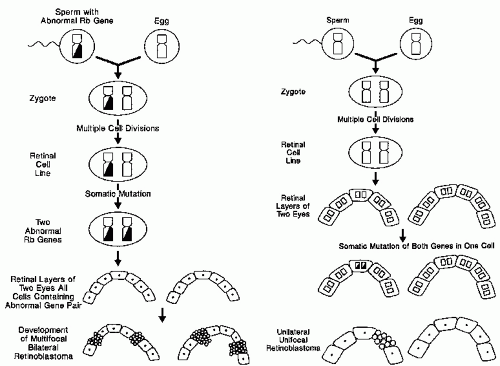
In its simplest statement, Knudson’s model suggests that children with sporadic RB are genetically normal at conception. During embryonic development, two somatic mutations (also called two genetic “hits”) occur in the cell line leading to the retinal photoreceptors. The resulting doubly mutated primordial retinal cell proliferates into RB tumors. In familial RB, the fertilized egg already carries one copy of the mutant gene (one hit). All descendants of this cell carry the mutation. If any cell sustains a somatic mutation (a second hit) to reach the doubly mutated state needed for tumor induction, RB develops. The two-hit hypothesis had the potential to explain both forms of RB (Fig. 5.2) (18,19,22,23).
Knudson’s hypothesis explained neither the precise gene affected nor the nature of the mutation or gene product that caused the malignancy. Evidence concerning the gene product came from the work of Cavenee and colleagues. They used DNA restriction fragment length polymorphisms (RFLPs) in the study of cancer. The cytogenic analysis was vital in uncovering the mechanism behind the two hits of RB. Investigators found that a few cases were associated with deletion of chromosomal band 13q14. Heterozygosities for linked but external markers of chromosome 13 were lost with the deletion but not with intragenic mutations. RFLPs supported the conclusion that either of these mechanisms can occur as a second event in RB. This work provided direct evidence for the identification of RB1 as a tumor suppressor gene (22,24,25,26).
What protein does the deleted 13q14 RB gene normally produce? How does it regulate cell growth? Why does its absence lead to malignancy? The search for the answers to these questions is at the heart of molecular cancer biology. In general, we may think of tumor formation as the possible result of mutations in two classes of genes: proto-oncogenes and tumor suppressor genes. When mutations occur in proto-oncogenes, which result in a gain of function and unbridled cell growth, they are called oncogenes. Proto-oncogenes participate in cell growth and proliferation via several mechanisms: the elaboration of cellular growth factors; the production and deployment of membrane growth factor receptors; intracellular signal transducers, which conduct growth-promoting signals from the membrane deep into the interior of the cell; and transcription factors that promote the ultimate production of proteins, which lead to cell proliferation. It is clear that proto-oncogenes have desirable physiologic functions in fetal development, normal childhood growth, wound healing, and desirable cell proliferation, as occurs in the aerodigestive tract, gut, and bone marrow. However, when proto-oncogenes go awry and become oncogenes, the persistent cell growth that results, without appropriate restraints of time and space, leads to cancer.
In contrast, tumor suppressor genes provide signals that constrain cell proliferation. Mutations in the tumor suppressor genes behave in a recessive manner at the molecular level. Consistent with Knudson’s hypothesis, when mutations occur in both alleles (two hits), the suppressive behavior of the gene is fully lost (i.e., only when both copies of the gene are inactivated by mutations does an abnormal phenotype, a malignant transformation, occur in a cell). A child born with one defective copy of a tumor suppressor gene has a predisposition to cancer. If the second copy of the gene is rendered inactive by a mutation, then the carcinogenic potential is realized. However, a child with two defective tumor suppressor genes has fully lost a constraint on cell growth, and cancer will ensue.
Figure 5.2 A: In heritable bilateral retinoblastoma, a gamete carrying a defective RB gene (the first hit) forms a zygote heterozygous at the RB locus. A later somatic mutation inactivates the other RB gene (the second hit), allowing bilateral multifocal tumors. B: In nonheritable retinoblastoma, inactivation of both genes in a retinal cell (two hits) by somatic mutations leads to the development of unilateral unifocal disease. (Modified from Toma NMG, Hungerford JL, Plowman PN, et al. External beam radiotherapy for retinoblastoma: II: lens sparing technique. Br J Ophthalmol. 1995;79: 112-117, with permission.)
The RB protein (pRB) is intimately involved in control of the cell cycle. A transcription factor called E2F (the name comes from its initial identification as being involved in the adenovirus E2 promoter) helps drive the cell through the cell cycle to mitosis. When pRB binds E2F, E2F is not free to activate promoters of DNA synthesis. In addition, the pRB-E2F complex downregulates the expression of many G1 exitpromoting genes such as c-myc and c-myb. In this way, pRB inhibits the cell from duplicating itself. If pRB is phosphorylated, the phosphorus groups displace the E2F from its binding site. Then the E2F launches into action and promotes cell division. If the pRB is absent or defective, then the factors leading to cell proliferation have no countervailing force.
The growth repressive action of RB occurs, in part, via the recruitment of chromatin-remodeling complexes to promote a region. These complexes mediate chromatin condensation and the subsequent inhibition of transcription. In addition to its role in self-cycle control, RB has been implicated in regulating a wide variety of cellular processes. These include DNA replication, differentiation, and apoptosis. RB potentially interacts with more than 100 different cellular proteins. The functional relevance of most of these interactions has not been elucidated.
A generation of pediatric radiation oncologists was taught that RB has an autosomal dominant inheritance pattern with an 80-90% penetrance (1,7). This is now known to be incorrect. RB is the result of an autosomal recessive pattern; only in the presence of damage to two alleles (two hits) will cancer develop. It must be the case that, in the presence of an inherited single defective allele, the occurrence of a mutation in the second allele is common; this gives the impression of dominant inheritance.
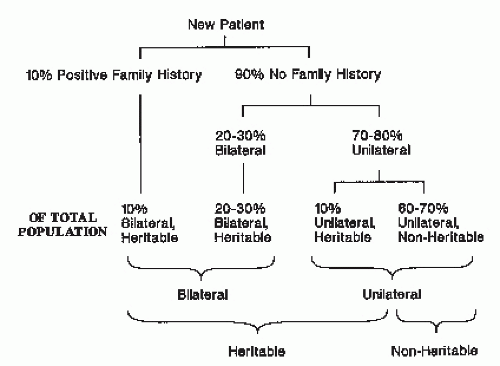
RB may be inherited from an affected parent or may be the result of sporadic mutations (26). Although most cases of RB are sporadic, 25-40% are familial (inherited from an affected parent who survived RB, a nonaffected gene carrier parent who has no clinical signs of RB, or a parent with a new germ-line mutation). Hereditary cases usually are bilateral and multifocal; they occur at a younger age than sporadic cases, which are, in comparison, more often unifocal and unilateral. Of all patients with newly diagnosed RB, about 10% have a positive family history, and most of these have bilateral disease. Of the 90% with no family history, 20-30% have hereditary, bilateral disease. The remaining 65-80% are unilateral; of these unilateral cases about 10% is hereditary and 90% nonhereditary (Fig. 5.3) (5).
There is also new information in which the effect of the human papilloma virus (HPV) has been questioned as a possible inducement agent for development of RB in both the sporadic and familial cases (27, 28, 29). These reviews have shown various HPV subtypes identified in RB tumor samples. The question remains as to whether they are part of the natural environment of these cases or, in fact, are agents for viral intervention for the development of RB.
WORKUP
RB commonly presents with a white pupillary light reflex (called leukocoria) (4,10,11). The parents may notice this abnormal appearance in a flash photograph (Fig. 5.4) because a photographic flash bounces light through the pupil and conjunctiva to produce a red appearance in a color snapshot. However, large RB, or an RB-producing retinal detachment, produces the white reflex when the flash bounces off it. This effect can also be seen with a handheld ophthalmoscope. A pediatrician or ophthalmologist doing a surveillance examination of a child with a positive family history or during the course of a routine examination may also discover RB.
Figure 5.3 The distribution of unilateral, bilateral, heritable, and nonheritable cases of retinoblastoma.
On physical examination one notes a raised white, white-yellow, or white-pink mass (2). Tortuous vessels may be seen feeding the tumor. Cells may break off from the main tumor mass and grow as small vitreous seeds (4). Because RB may be multifocal, it is necessary to examine the entire retinal surface, generally with the patient under anesthesia.
When RB presents as a mass, the differential diagnosis includes astrocytic hamartoma, Toxocara canis granuloma, the infected emboli of subacute bacterial endocarditis or toxoplasmosis, and other types of severe uveitis. When RB causes retinal detachment, the differential diagnosis includes Coats disease, retrolental fibroplasia, and persistent hyperplastic vitreous (2,3, 4, 5,7). Biopsy of a suspected RB or vitreous aspiration for enzyme studies is generally felt to be contraindicated because of the risk of choroidal seeding (4). The consequences of intraocular procedures in patients with RB are discussed later in this chapter.
Figure 5.4 Leukocoria in a flash photograph of a 6-month-old girl with retinoblastoma. Over time, the eye improved, and irradiated with 43.6 Gy, she retained excellent vision at 20-year follow-up. (Reproduced with permission of the patient’s parents.)
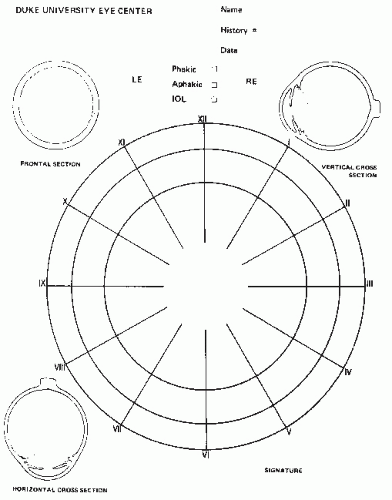
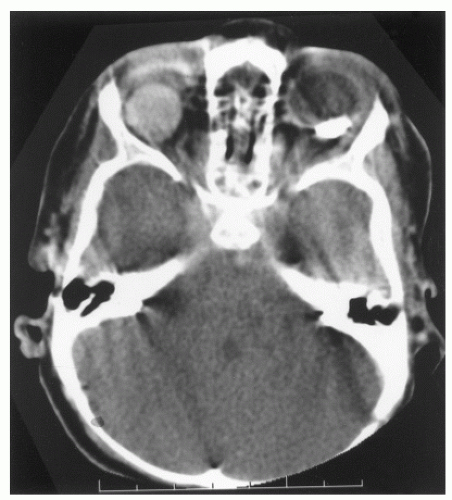
Retinal drawings and photographs, along with a written description, are used to record whether single or multifocal tumors are present (Fig. 5.5). Ultrasound is also useful for documenting tumor location and size. The distance from the cornea to the back of the lens can also be measured with ultrasound to aid in lateral field radiotherapy planning. Computed tomography (CT) scan is effective in demonstrating tumor calcification. A cranial CT scan may accompany the orbital study to assess the presence of intracranial extension of the primary tumor and the possibility of a synchronous pineal tumor.
Among the tests used to detect metastatic disease are a lumbar puncture with cerebrospinal fluid (CSF) cytology and a bone marrow biopsy and aspirate. Pratt et al. (30) reported no positive CSF cytologies or bone marrow aspirates in 109 children with RB confined to the globe. The only positive studies were in patients with extraocular tumor extension (i.e., through the sclera or beyond the cut end of the optic nerve) or with bone, distant soft tissue, or brain metastases. These data are supported by other reports (31,32). In modern practice, routine lumbar puncture and bone marrow aspiration are not justified for RB confined to the retina without optic nerve involvement or other suggestion of extraocular extension. In the presence of symptoms suggestive of metastatic disease, a bone scan and plain bone films are indicated.
PROGNOSTIC FACTORS AND STAGING
The staging system for RB must fulfill at least two requirements. First, it must predict the likelihood of cure, a requirement of all malignancy staging systems. However, an important goal of RB treatment is preservation of sight in the affected eye. To this end, some eyes are treated with chemotherapy, cryotherapy, laser photocoagulation, hyperthermia, radioactive plaque, and external beam radiotherapy as an alternative to enucleation. Therefore, the second requirement for staging is to predict the likelihood of visual preservation.
Figure 5.5 A standard retinal drawing used to localize intraocular tumors. The drawing represents the inner curved surface of the eye. The space between the outermost circle and the middle circle represents the pars plana. The middle circle is the ora serrata, the anterior termination of the retina. The most inner circle is the geometric equator of the globe. The central area of the drawing indicates the macula, the yellow spot in the center of the retina. The macula contains a pit, the fovea centralis, where closely packed cones function as the area of most acute vision. The optic nerve’s exit (the optic nerve head or disc) is 2 mm medial to the macula. The disc is approximately 1.5 mm in diameter and is often used as a measure of tumor dimensions.
The most widely used grouping system for RB was proposed by Algernon Reese (5) and Robert Ellsworth (Table 5.1) (33). This system does not predict survival probability well. However, it does predict the chance of visual preservation with conservative therapy. At least two staging systems have attempted to predict prognosis for survival and include information on disease extension beyond the globe (8,30). Of these systems, the St. Jude Children’s Research Hospital (SJCRH) system has been used somewhat more frequently. The most recent clinical protocols for combined-modality RB therapy use a new system based on the work of the Children’s Oncology Group (COG) and Murphee et al. of Children’s Hospital of Los Angeles. This staging system is gaining increasing popularity (Table 5.2). It attempts to assess viability and outcomes related to chemotherapy administration.
Table 5.1 The Reese-Ellsworth System of Classifying retinoblastoma
Group I: Very Favorable
A.
Solitary tumor, less than 4 DDa in diameter, at or behind the equator
B.
Multiple tumors, none larger than 4 DD in diameter, all at or behind the equator
Group II: Favorable
A.
Solitary tumor, 4-10 DD in diameter, at or behind the equator
B.
Multiple tumors, 4-10 DD in diameter, all at or behind the equator
Group III: Doubtful
A.
Any lesion anterior to the equator
B.
Solitary tumors, larger than 10 DD in diameter, behind the equator
Group IV: Unfavorable
A.
Multiple tumors, some larger than 10 DD
B.
Any lesion extending anterior to the ora serrata
Group V: Very Unfavorable
A.
Massive tumors involving more than half of the retina
B.
Vitreous seeding
a The optic nerve’s exit (the optic nerve head or disc) is approximately 1.5 mm in diameter. The disc diameter (DD) often is used as a measure of tumor dimensions.
There is also a system that addresses intraocular and extraocular RB described by Grabowski and Abramson.
HISTOLOGIC EVALUATION OF RETINOBLASTOMA
RB is a poorly differentiated malignant neuroectodermal tumor. There are often significant necrotic and apoptotic changes. Although the cytologic details of the tumor may vary from case to case, the cell type, the degree of differentiation, and necrosis have not been defined in themselves as risk factors for local recurrence or metastasis (35). Some have asserted that poorly differentiated tumors are more likely to respond to radiation, but this information is not available at the time of radiation treatment. Some investigators believe that there is an association between vascular density and prognosis in RB. This may merit further investigation.
Classically, four growth patterns are recognized in RB:
Endophytic RB, which grows from the retina toward the vitreous, appears to be a mass protruding into the vitreous chamber. These often friable and necrotic tumors may produce small clusters of tumor cells that are detached from the main mass and form satellite tumor nodules. These can range from localized tumor nodules within the vitreous, known as vitreous seeding, up to diffuse involvement, which some call the snowstorm effect.
Exophytic RB typically grows from the outer retinal layers and extends beneath the detached retina toward the choroid. Dislodged masses may implant on the retinal pigment epithelium and erode through the Bruch membrane into the choroid.
Diffuse plaque-like RB defies common morphologic patterns. It grows diffusely and insidiously in the retina without forming a detectable mass. Such a growth pattern can present a confusing clinical picture.
A mixed endophytic and exophytic pattern may also occur (36).
Table 5.2 ABC Classification for Intraocular Retinoblastoma
Group A
Small tumor confined to the retina
No tumor greater than 3 mm in diameter
No tumor less than 2 DD (3 mm) from fovea or 1 DD (1.5 mm) from optic nerve
No vitreous seeding
No RD
Group B
Tumors confined to the retina, any location
No vitreous seeding
No retinal detachment more than 5 mm from tumor base
Group C
Fine diffuse or localized vitreous or subretinal seeding
No tumor masses, clumps, or snowballs in the vitreous or subretinal space
More than 5 mm RD to total RD
Group D
Massive vitreous or subretinal seeding
Snowballs or tumor masses in the vitreous or subretinal space
More than 5 mm RD to total RD
Group E
No visual potential or presence of one or more of the following
Tumor in the anterior segment
Tumor in or on the ciliary body
Neovascular glaucoma
Vitreous hemorrhage obscuring the tumor or significant hyphema
Phthisical or prephthisical eye
Orbital cellulitis-like presentation
DD, disc diameter; RD, retinal detachment.
RISK FACTORS FOR METASTASES IN RETINOBLASTOMA THAT CAN BE IDENTIFIED AT THE TIME OF ENUCLEATION
The ocular pathologist plays a crucial role in determining whether RB has a risk of dissemination because it has broken out of its ocular confines. Reported risk factors include the following:
Optic nerve invasion: The presence of tumor beyond the lamina cribrosa is a risk factor for metastatic disease because access to the subarachnoid space allows RB cells to spread to the spinal fluid and central nervous system. Optic nerve invasion is present in approximately one third of nucleated globes and often correlates the presence of choroidal or scleral extension. Optic nerve invasion is categorized as preliminary, laminar, postlaminar, and up to the line of transection. The latter occurrence is quite rare. In a study of 261 cases, the multivariate risk of metastasis was 9 when the optic nerve was involved up to the resection line, compared with 4 when the RB extended retrolaminar but not up to the resection line. A study of 172 patients found that the disease-free survival rate was 97% when the optic nerve was not involved and 55% when it was involved up to the line of resection. The prognosis was intermediate with retrolaminar involvement. When patients with extrascleral extension and optic nerve involvement up to the resection line were excluded, retrolaminar involvement still was a significant prognostic factor for metastasis (37).
Uveal invasion: Choroidal invasion has long been considered an important risk factor for metastatic disease because it allows the tumor access the scleral and emissary veins. However, choroidal invasion is difficult to quantify. In a study from Wills Eye Hospital, it appeared that patients with choroidal invasion were more likely to develop distant metastasis than those without. However, when patients with optic nerve invasion were excluded, there was no longer a significant risk, although there was still a trend toward the metastasis with choroidal invasion (p = 0.1) (36).
Orbital invasion: When tumor extends to the sclera, it gains access to the vascular and lymphatic channels outside the eye. This creates a significant risk for metastatic disease and is generally considered an indication for more aggressive local and systemic treatment. Microscopic orbital extension often is impossible to diagnose by clinical examination. Preoperatively, larger extensions may be imaged by ultrasound, CT, or magnetic resonance imaging (MRI).
Choroidal involvement: Invasion of the choroid usually is considered a poor prognostic sign. It is almost certainly true that hematogenous spread of RB occurs via choroidal vessels. However, choroidal invasion is not rare; it occurs in up to 62% of cases. Distant metastases are rare. It seems that the prognostic correlation is less with choroidal invasion alone than with the volume of choroidal invasion and its correlation with other risk factors.
Involvement of the optic nerve beyond the lamina cribrosa is a risk factor for orbital recurrence and central nervous system (CNS) dissemination. Even worse is the involvement of the distal cut end of the optic nerve with tumor (35). Tumor involvement of the scleral and emissary veins and episcleral tissues also forebodes a poor prognosis. Multivariate analysis of data from 330 children at St. Bartholomew’s Hospital and Moorfield’s Eye Hospital showed that the deleterious effect of extensive choroidal invasion as a prognostic factor was lost in the absence of retrolaminar invasion or invasion of the cut end of the optic nerve. Two patient groups had a particularly poor prognosis: retrolaminar extension and extensive choroidal invasion (5-year survival, 31%); and extensive choroidal invasion and invasion of the cut end of the optic nerve (5-ear survival, 25%) (4). In 62 children only 1 death was attributable to metastatic disease from choroidal invasion alone (38,39).
SELECTION OF THERAPY
The primary goal of RB therapy is cure. Because RB infrequently metastasizes, the chance of cure is excellent. However, this must be viewed with the knowledge of otential deleterious effects of the local therapy, which in its worst scenario includes development of second malignant tumors.
The actuarial overall 5-year survival rate for 731 children with RB seen at St. Bartholomew’s Hospital and Moorfield’s Eye Hospital from 1960 to 1988 was 87% (4). The 50-month actuarial overall survival of 52 SJCRH patients with initial intraocular disease was 97% (8).
Therefore, it is appropriate that a secondary goal of treatment is preservation of vision in the affected eye. Enucleation is recommended only in a blind eye or an eye for which there is no reasonable expectation of sight after cryotherapy, phototherapy, or radiotherapy. Every effort should be made to preserve vision in a sighted eye. This rule pertains to both bilateral and unilateral diseases. Often, however, children with unilateral disease have locally advanced tumors with little hope of vision, so many of these patients undergo enucleation. In bilateral disease, enucleation of the more severely affected eye is indicated only if the eye is blind. If both eyes are sighted, then an effort should be made to preserve both eyes.
Enucleation
In an enucleation for RB, anterior traction is placed on the globe after the rectus muscles are severed. The optic nerve is then cut near its exit from the socket. Obtaining a long segment of nerve is important if the tumor is within the nerve. In young children, orbital growth slows after enucleation. As the child grows, the orbit appears small. This may be ameliorated by a properly fitting orbital prosthesis.
As we have gained knowledge in the treatment of RB, there has been a noted reduction in the use of enucleation for both unilateral and bilateral cases. (36, 37, 38).
Enucleation is indicated in unilateral RB, where the eye is blind. In bilateral RB when both eyes are blind, a bilateral enucleation is done. If one eye is blind, a unilateral enucleation is done. It is also recommended when local recurrence of tumor can no longer be controlled with more conservative measures (13,40) (Fig. 5.6).
Exenteration
An exenteration is the removal of the globe, extraocular muscles, lids, nerves, and orbital fat. Blood loss may be significant. In the opinion of some ophthalmologists, the indications for exenteration in RB include extensive local tumor breaching the globe (exenteration in this situation generally is followed by postoperative radiotherapy and chemotherapy) and recurrence of tumor in the socket after enucleation. However, some cases of local recurrence may be locally managed with external beam radiotherapy, a radioactive implant or a radioactive mold, and chemotherapy.
Local Therapy
The four local therapies for RB are cryotherapy, photocoagulation, laser hyperthermia, and radioactive plaque applications.
Until the early 1990s, in children at high risk for multicentric disease, local therapy was thought to be appropriate only when there could be serial follow-up with repeated local therapy or with external beam radiotherapy when necessary. In children with bilateral disease, an average of five tumor foci are randomly distributed (2,41). These foci may not appear simultaneously but may occur months after successful local treatment of the sentinel lesion, sometimes as long as 60 months after the initial index lesion. In children with multifocal disease or in those at high risk of developing multifocal disease, as well as in children with large tumors, it was believed that local therapy generally should not be used.
Figure 5.6 Bilateral retinoblastoma treated with enucleation on one side with a prosthesis in place. A calcified macular lesion is seen in the remaining eye.
More recently, thinking concerning local therapy is very different. With the increase in use of systemic chemotherapy, local therapy is now used in multifocal disease. Local therapy may be interspersed with chemotherapy or may follow it. Programs using focal therapy in this way are reviewed later in this chapter.
Photocoagulation
The technique of photocoagulation is based on obliteration of the retinal vessels. With the child under anesthesia, a white retinal burn, surrounding the tumor by 1 mm, is painted with the laser beam. Special attention is directed to closing feeding vessels (42). The tumor is encircled by the burn and regression depends on interruption of blood supply. Direct photocoagulation of RB should be avoided because small explosions can release viable tumor cells into the vitreous and lead to tumor recurrence (4). In primary therapy, photocoagulation may be used for tumors up to 4.5 mm at the base and up to 2.5 mm thick if they were not close to the macula or disc, where retinal damage would generate a large scotoma. Vitreous seeding is a contraindication (11,43). Photocoagulation may be used for small tumor recurrences after irradiation to avoid the risks of reirradiation. With proper case selection, photocoagulation has a local tumor control probability of about 70% (42).
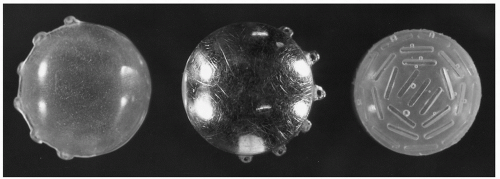
Figure 5.7 Equipment for 125I ocular plaque construction and placement includes a dummy plaque to aid in the placement of the necessary retention sutures (left), a gold backing with lug holes for sutures (center), and a plastic insert to hold the radioactive 125I seeds (right).
Laser Hyperthermia
Laser hyperthermia is generated by a diode laser (810 nm) on continuous mode. A single spot, 0.8-2.0 mm in diameter, is placed on the center of the tumor using the aiming beam. An output of 300-700 mW is selected based on the tumor size. Tumors are heated for 10-30 minutes per session. It is estimated that a central tumor temperature of about 46°C is reached. As the heat disperses through the tumor, the temperature decreases about 2°C for each millimeter beyond the treatment spot. In this technique the heat is used principally to enhance the binding and cytotoxicity of platinum drugs, a method supported by experiments using rabbit ocular melanoma (42,44,45). Whole eye hyperthermia in combination with external beam irradiation has been shown to be effective in controlling murine transgenic RB but to our knowledge has not been used in humans (46).
Cryotherapy
With cryotherapy a tumor is localized and indented, transsclerally, with a nitrous oxide cryoprobe. The freez (80°C) is then applied until the tumor is completely covered with a frozen vitreous. The freeze-thaw cycle is repeated at least three times (41). Cryotherapy is indicated for the primary therapy of RB in small tumors anterior to the equator, without vitreous seeding, which can be reached with the cryoprobe (posterior tumors are difficult to reach, and the risks of freezing the macula or nerve increase); in local recurrence or tumor persistence after irradiation; and in conjunction with chemotherapy (44).
Cryotherapy can induce acute retinal edema and accumulation of subretinal fluid. To avoid retinal detachment, some ophthalmologists use the laser to create a retinal barrier to fluid leakage. Disruption of the retina by cryotherapy may increase intravitreal penetration of systemic carboplatin.
Radioactive Plaque Application
Plaques are used for solitary 2- to 16-mm basal diameter unilateral lesions located more than 3 mm from the optic disc or fovea, generally less than 10 mm thick, for two lesions that are small enough or close enough to be covered by one plaque, and for local failure after other therapies (Fig. 5.7). Plaques can be used if there is a small amount of vitreous seeding over the tumor apex (10,47, 48, 49, 50).
Before the operative procedure, the tumor’s maximum basal diameter and maximum height are ascertained by physical examination and ultrasonography. In treatment planning, it is customary to allow 1 mm for scleral thickness, although there is some normal variation in this measurement (51). The operative procedure begins with a careful eye examination using magnifying lenses. After confirming the tumor anatomy, the surgeon opens the conjunctiva around the periphery of the limbus (a peritomy). Muscle hooks are used to snare rectus muscles and rotate the eye. Traction sutures sometimes are used. It may be necessary to disinsert a muscle in order to visualize the tumor. With the room darkened, a transilluminator is placed over the pupil. The shadow cast by the tumor is marked on the sclera with a marking pen or with electrocautery. Tumors that cannot be transilluminated are located by ultrasound. A clear dummy plaque is then brought into the operative field. Allowance for a 2 mm of margin on either side of the basal diameter is standard. A dummy plaque, which is an exact copy of the to-be-used radioactive plaque, is used to assure correct coverage; it is also used to assist in the initial retention suture placement, thus reducing exposure to the surgical team. The “live” plaque is substituted and the retention sutures are tightened down, and the eye is rotated back into place. The conjunctiva is then closed. The patient generally remains hospitalized for the duration of the application. The plaque is then removed after the appropriate dose is delivered (52).
Several plaque types are available. The 60Co plaque (1.17 and 1.33 MeV, half-life 5.2 years) may be purchased in a circular or crescentic configuration to fit around the optic nerve. The 125I plaque (27-35 keV, half-life 60 days) with lip consists of 125I seeds glued in a carrier within a gold shield. These plaques are available in a circular or notched configuration. 192Ir (295-612 keV, half-life 74.5 days) and 109Ru (beta emitter) plaques are also available (53).
Each of the four available plaques (60Co, 125I, 192Ir, and 109Ru) has advantages and disadvantages. 60Co plaques may be purchased and assembled in standard sizes. The long half-life means that the plaque may be used for several years before the treatment times become unacceptably long. The high-energy 60Co and the breadth of the high-dose isodose curves mean that thick and infiltrative tumors can be treated. However, shielding of periocular normal tissues behind the plaque is impossible. Effective shielding on the back of the plaque is achievable with 125I and 109Ru. Shielding from the gold lip on the 125I plaque necessitates expert placement lest tumor be missed. 125I plaques must be assembled for each case; although this involves extra work, it allows individualization of the plaque. Hospital personnel exposure is minimal with 125I or 109Ru. A shielded 125I plaque with a lead eye patch allows the nursing staff and the child’s parents to provide care while observing appropriate radiation safety precautions.
Using 60Co plaques, Stallard administered 40 Gy to the tumor apex in 1 week. Of 69 children with tumor involving one fourth of the retinal area or less, 63 were successfully treated with a plaque. When the tumor involved one fourth to one half of the retinal area, success was achieved in 8 of 10 cases (54).
Among the best-characterized clinical series of plaque, brachytherapy for RB is that of the Wills Eye Hospital of Philadelphia (49,50,53). This series describes the outcomes for more than 140 children with RB treated between 1976 and 1999. In these children, 148 tumors were treated with radioactive plaques for recurrent or residual RB after other therapies failed, and 60 tumors were treated primarily. The median tumor dosage at the tumor apex was 40 Gy, with 125I used in about 90% of all plaques. With a median follow-up of 34 months, recurrences developed in 17% and 20% of cases at 1 and 4 years, respectively. In tumors treated with a radioactive plaque as primary therapy, recurrences were observed in 12% and 20%. Metastases developed in only four patients, all with Reese-Ellsworth group Vb tumors. On multivariate analysis, the risk of recurrence was increased significantly by the presence of vitreous or retinal seeds and by increasing age at diagnosis. In earlier studies, visual acuity of 20/400 or better was obtained in 62%. A series on a subject of patients treated with chemoreduction (CRD) and who subsequently failed and were treated with 125I plaque reported a 95 % 5 year local control rate in 59 eyes treated initially with CRD and 100 % 5 year local control in 25 initially receiving CRD and external beam irradiation.
In another recent series, 106Ru was utilized in 41 eyes. It was used as first- and second-line therapy in 16 and as salvage in the remaining. Of 41 patients, 31 have eye retention. Complications included cataract (9.7%), retinopathy (2.4%), and retinal detachment (17%), again showing effectiveness in controlling relapsed RB (55).
In the past, it was reported that for large tumors with vitreous seeds, multiple sequential plaques (rotating plaques) could be used. In this technique, two plaques are applied in one operation to opposite quadrants of the eye. In a second operation, these two plaques are rotated to the remaining quadrants. In a third operation, the plaques are removed. This technique delivers about 40 Gy to the midvitreous and 160 Gy to the sclera. Initial reports indicated that useful vision was retained in 14 of 16 patients treated with plaques as the single irradiation modality. Useful vision was retained in 22 of 36 patients plaqued after failure of some other treatment (10,49,52). Later reports showed a significant incidence of radiation retinopathy with this technique, and it has been largely abandoned (52).
In carefully selected patients with small primary RB or for recurrent tumors after other treatment, a radioactive single-plaque application of 30-40 Gy to the tumor apex is reasonable therapy. With growth in the use of chemotherapy as primary treatment for RB, plaque therapy has joined photocoagulation, laser treatment, and cryotherapy as an adjuvant focal treatment after or interspersed with chemotherapy. When a plaque is used in a child who has also received chemotherapy, many believe that 40 Gy as an apical dosage is too high. The occurrence of postbrachytherapy retinitis has led to a dosage reduction to 25-30 Gy to the tumor apex. Plaques have little chance of producing orbital bone hypoplasia and should not contribute to the risk of orbital bone sarcoma. Long-term complications of plaque radiotherapy include retinopathy, cataracts, maculopathy, papillopathy, and glaucoma (49,54). In the Wills Eye Hospital series of more than 140 children treated with plaque radiotherapy, only one second cancer was observed in the field of plaque irradiation over a mean follow-up of 49 months.
External Beam Radiotherapy
When RB is multifocal or close to the macula or optic nerve with preservation of vision, it has been found that cryotherapy, photocoagulation, or plaque therapy is not adequate and that enucleation is too drastic. In such situations, which are quite common, external beam irradiation or chemotherapy with focal therapy is used. These types of therapy are also indicated for large tumors and vitreous seeding. Hilgartner (56) reported treatment of a case of bilateral RB with x-rays in 1910. Verhoeff cured a case of RB with x-ray treatment in 1918. The patient died in 1972 with tumor controlled (57). Historically, the Reese-Ellsworth grouping system has been used to predict the probability of success for external beam irradiation (Table 5.1). Large lesions tend to fail after teletherapy. Some say that anterior lesions are also more likely to fail after teletherapy, but the risk of failure of anterior lesions probably was related to the practice, by some radiotherapists, of treating with a lateral beam with the anterior field edge at the rim of the bony orbit. This technique underdoses the anterior globe (8,58, 59, 60, 61). Technical factors play a large role in the probability of success of external beam therapy and the frequency of complications.
Technique
The goals of conventional external beam radiotherapy are to provide a homogeneous and tumoricidal dose to the retinal anlage and vitreous and to respect tolerance of normal tissue structures. At least five arguments have been put forward in support of this expansive view of treatment volume. First, in many cases RB represents a field change in which all retinal cells have a genetic neoplastic potential; therefore, the entire retina must be treated. Second, vitreous seeding can occur. Third, multiple tumors may arise from a primary RB. Fourth, the tumor may spread via the subretinal space. Finally, retinal differentiation progresses from posterior to anterior and from superior to inferior. Subclinical disease may exist in the immature retina and must be included in the treatment (15). The argument in favor of a more restrictive tumor volume is that selected cases will be unilateral and unifocal and therefore amenable to more focal irradiation, as might be delivered to a fixed target with protons.
Only gold members can continue reading. Log In or Register to continue

 (80°C) is then applied until the tumor is completely covered with a frozen vitreous. The freeze-thaw cycle is repeated at least three times (41). Cryotherapy is indicated for the primary therapy of RB in small tumors anterior to the equator, without vitreous seeding, which can be reached with the cryoprobe (posterior tumors are difficult to reach, and the risks of freezing the macula or nerve increase); in local recurrence or tumor persistence after irradiation; and in conjunction with chemotherapy (44).
(80°C) is then applied until the tumor is completely covered with a frozen vitreous. The freeze-thaw cycle is repeated at least three times (41). Cryotherapy is indicated for the primary therapy of RB in small tumors anterior to the equator, without vitreous seeding, which can be reached with the cryoprobe (posterior tumors are difficult to reach, and the risks of freezing the macula or nerve increase); in local recurrence or tumor persistence after irradiation; and in conjunction with chemotherapy (44).