© Springer International Publishing AG 2017
Pietro Ruggieri, Andrea Angelini, Daniel Vanel and Piero Picci (eds.)Tumors of the Sacrum10.1007/978-3-319-51202-0_1212. Schwannoma of the Sacrum
(1)
First Department of Orthopaedics, National and Kapodistrian University of Athens, Athens, Greece
(2)
Department of Orthopedics and Orthopedic Oncology, University of Padova, Padova, Italy
12.1 Introduction
Nerve sheath tumors comprise only a small portion of the wide variety of lesions that occur in the sacrum [1–5]. Schwannomas (neurilemomas) are benign, slow-growing neurogenic tumors arising from Schwann cells of the peripheral nerve sheath. Sacral schwannomas are exceedingly rare, with merely around 50 cases reported in related literature [6]. Due to their largely indolent nature, the mobility of the sacral nerve roots, and the generous width of the sacral canal, sacral schwannomas frequently grow to a considerable size prior to detection; hence, giant sacral schwannomas are often described in related reports [7, 8]. Although benign, these tumors may occasionally pose a surgical challenge, as en bloc resection may be associated with disproportionally high surgical morbidity [9].
12.2 Epidemiology
Sacral schwannomas are decisively more rare than the common spinal schwannomas [6], accounting for less than 1–5% of all spinal schwannomas [6]. Schwannomas usually occur in individuals between 20 and 50 years of age with no gender predilection; they are associated with von Recklinghausen’s disease (neurofibromatosis type II) in 18% of cases [10]. Schwannoma can involve the sacrum in one of three ways: (1) secondary erosion by an extraosseous tumor; (2) tumor arising from a nerve coursing through a canal and causing erosion of the bone, creating a dumbbell-shaped configuration; or (3) tumor arising centrally (intramedullary or intraosseous) [11]. The first two mechanisms are more common. Intraosseous schwannomas are exceptional, accounting for less than 0.2% of all primary bone tumors, with fewer than 200 cases described in literature, in all locations [12–21]. Intraosseous schwannomas have also been described in a setting of neurofibromatosis type III (schwannomatosis) [21]. Giant intraosseous sacral schwannomas were first reported by De Santo et al. in 1940 [17], and so far, approximately 50 cases have been reported [6, 18, 19].
12.3 Presentation and Diagnosis
Most patients with a sacral schwannoma remain asymptomatic for a long period of time. If present, symptoms are generally mild, and it is not until the tumor becomes very large that patients notice pain or swelling [9, 19]. In a report, a patient was harboring a sacral schwannoma for more than 25 years before the onset of symptoms [22]. The mean duration of symptoms prior to diagnosis ranges from 1 to 7 years [18, 20, 23, 24]. Pain is the most common presenting symptom. It may be either localized, low-back, or radicular pain extending in the distribution of one or more lumbosacral nerve roots. It may also be associated with sensory defects, such as paresthesias or dysesthesias. Urinary hesitancy and retention, weak stream and overflow, or unconscious incontinence may be present if bladder innervation is compromised. Patients may also present with recurrent bouts of cystitis [6]. Complaints of constipation or an unpleasant feeling of rectal fullness or pain (tenesmus) are alternative modes of presentation [6, 9, 23]. Potential physical signs previously described include lower extremity muscle weakness, atrophy, decreased deep tendon reflexes (particularly the Achilles tendon reflex), as well as diminished saddle and lower extremity sensation [9, 19].
Rectal examination is an important diagnostic maneuver in the setting of a suspected sacral tumor. The mass is often palpable, permitting a further restriction of differential diagnosis [24]. Neurogenic tumors are usually soft, firm, and off the midline, whereas chordomas are often solid and irregular tumors occurring in the midline of the axial skeleton [9]. A decisive aid in the definitive diagnosis of a sacral schwannoma is biopsy. However, its use has been somewhat controversial. Some authors sustain that biopsy is unreliable for the diagnosis of giant nerve sheath tumors because of the secondary degenerative changes frequently present and underline the potential risk of complications associated with the procedure, such as infection or bleeding [18, 25–27]. Others, however, believe that biopsy is essential for determining the treatment of choice or surgery type, as well as to exclude some tumors that would not benefit from a surgical procedure, such as lymphoma [28–30]. In the latter case, biopsy should be performed with oncological principles. The biopsy tract should be marked for possible resection, if pathology results yield the presence of a malignant tumor, such as a sacral chordoma.
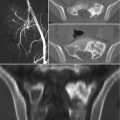
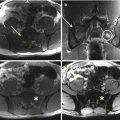
12.4 Imaging
In radiographs, the lesions are lytic, with a narrow sclerotic margin usually present at the periphery. The bone contour may be expanded, but there is no periosteal new bone formation. The cortex may be thinned or partially eroded, but no true cortical destruction or extension into soft tissue is usually seen. Central calcification or ossification is absent [31]. However, radiography is almost always nonspecific, and the overlying bowel shadow may frequently obscure bony details. Therefore, CT and MR imaging provide a more complete and detailed assessment of the sacrum, also demonstrating the relationship of the tumor with the anatomical structures present within the pelvis. CT is superior in demonstrating bony detail, whereas MR imaging allows for a better contrast for the study of soft tissue and provides a better display in multiplanar views [8, 32]. On MR imaging, schwannomas are isointense to muscle on T1-weighted and hyperintense on T2-weighted images. After administration of contrast medium, central enhancement is typical [6]. In contrast, malignant peripheral nerve sheath tumors tend to have content with heterogeneous contrast enhancement [6]. Si et al. [8] performed a retrospective study on 49 patients with sacral tumors aiming to define CT and MR imaging features that can safely differentiate between sacral chordoma, sacral giant cell tumor, and giant sacral schwannoma. According to the authors, middle age, a midline and lower sacral location with irregular and fuzzy bone residues, fresh bleeding, and ascending extension in the sacral canal are characteristic features of sacral chordoma; in contrast, younger age, eccentric or upper sacral location, incomplete bony shell, polycystic areas, fluid-fluid levels, and involvement of the sacroiliac joints are features pointing toward a giant cell tumor; finally, pressure bone erosion instead of bone destruction, large and central cystic areas, and absence of adjacent muscle or sacroiliac joint involvement are more likely to represent a giant sacral schwannoma [8].
12.5 Pathology
Schwannomas are composed entirely of cells with the immunophenotype and ultrastructural features of Schwann cells. Macroscopically, intraosseous schwannomas are soft, tan-gray lesions with sparse, yellowish, patchy areas. Cystic and myxoid changes, as well as bleeding, are frequently seen. When the lesion is located in a neural canal or foramen, a capsule may be discernible [11]. The microscopic features of intraosseous schwannoma are similar to those of their more common soft tissue counterparts [14]. There are, however, numerous schwannoma variants. In their classic form, the tumors have two basic patterns: hypercellular or dense (Antoni A) and hypocellular or loose (Antoni B). In Antoni A areas, schwannoma cells are compactly arranged to form focally palisading structures (Verocay bodies). In Antoni B areas, schwannoma cells are widely separated by a loose intervening collagenous matrix. Lesions may exhibit some nuclear atypia, with occasional mitoses. Long-standing schwannomas often show stromal and vascular degenerative changes including central tissue loss with cystic formation, necrosis without nuclear palisading, nuclear atypia, widespread hyalinization, and calcification [33]. Cellular schwannomas differ from the classic type in that the dense Antoni A pattern comprises 90% or more of the tumor area with a more uniform pattern, a lack of Verocay bodies, and a frequent lymphocytic infiltration [29]. A sacral melanocytic schwannoma has only been reported once [34]. Differential diagnosis should include neurofibroma and malignant nerve sheath tumors. Neurofibroma is another benign nerve sheath tumor that rarely involves the sacral region. It is composed of cells with a polymorphic cellular phenotype. These include Schwann and perineural cells as well as endoneurial fibroblasts [35]. Malignant nerve sheath tumors, such as malignant schwannoma or neurofibrosarcoma, very rarely occur in the sacrum, as well. Intrafascicular spread, local invasion, a fibrous pseudocapsule, gross areas of necrosis, numerous mitoses, and pleomorphism are hallmark features. Previous local radiotherapy may be a contributing factor to the development of malignant nerve sheath tumors [36, 37].
Immunohistochemically, tumor cells are positive for vimentin and S-100 protein. Differential diagnosis should include desmoplastic fibroma, well-differentiated fibrosarcoma, fibrous dysplasia, and nonossifying fibroma. Schwannoma should also be distinguished from a malignant spindle cell neoplasm [11]. Cytogenetic analysis of schwannomas has determined the occurrence of a complete or partial loss of chromosome 22, which bears the tumor suppressor gene NF2, as the most frequent abnormality [38].
12.6 Treatment
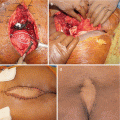
The mainstay of treatment for the patients with Schwannomas is surgical excision. Given the benign behavior of the tumors, excision can be accomplished by enucleation or curettage. However, sacral excision is usually difficult due to large size, robust blood supply, and frequent proximity to critical structures [9]. Furthermore, available studies suggest that intralesional curettage alone is probably unacceptable as it has a significantly high recurrence rate. In a series published by Abernathy et al. [18], 13 patients with sacral schwannoma were treated with intralesional excision; their local recurrence rate was reported to be as high as 54%, with four patients ultimately requiring reoperation. This data underscores the need for a more aggressive approach, more similar to that employed for malignant sacral tumors. Thus, most authors recommend en bloc resection as the most appropriate procedure for sacral schwannomas [17–19, 39].
An important aspect of the surgical treatment of sacral schwannomas is the choice of the surgical approach needed for efficient en bloc resection. This is often dictated by the exact anatomical distribution of the tumor, with emphasis on the amount of intrasacral and intrapelvic extension of the lesion. According to Abernathy et al. [18], tumor extension can occur in three directions: (1) in a cephalad direction into the spinal canal, often intradurally; (2) track through the sacral foramina; or (3) erode through the walls of the sacrum. Based on the anatomic extension of the tumor, Klimo et al. [9] proposed a classification of sacral nerve sheath tumors in an attempt to aid the selection of the appropriate surgical approach. According to their work, a type I sacral nerve sheath tumor is one that is confined to the sacrum; a type II (most common) has eroded either the anterior or posterior sacral wall and spread to adjacent spaces; and a type III is confined to the presacral space. They proposed the use of a posterior approach for type I, an anterior approach for type III, and a combined anterior-posterior approach for type II tumors. In a similar fashion, based on an extensive series of 48 patients who underwent surgery for sacral neurogenic tumors, Wei et al. [40] proposed an extended version of the previously mentioned classification system. According to this revised scheme, tumors are classified into four types with respect to tumor growth patterns. Type I tumors are confined to the intrasacral canal and manifest as an enlarged and swollen sacral canal; type II tumors extend through the intrasacral foramina into the presacral space, typically forming a giant presacral mass; type III tumors grow toward both the presacral area and the posterior subcutaneous tissue, forming a mass both anterior and posterior to the sacrum; and type IV tumors are confined to the presacral space, with no tumor observed within the sacral canal. For type I, as well as type II and type III cases in which the tumor extends below S1, the authors employ a posterior approach. For type II and III cases, in which the tumor expanded higher than S1, a combined anterior-posterior approach is recommended. Finally, for type IV cases, the authors propose a single anterior approach [40].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


