Fig. 1
Confocal micrographs of parasites stained with Fluo-4. Fluo-4 signal is redistributed to the parasite cytosol when the digestive vacuole is breached, resulting in an increased area of fluorescence. Arrowheads denote the hemozoin crystal, which is localized within the digestive vacuole. Scale bars represent 5 μm
2 Materials
2.1 Parasite Culture
1.
Culture media: RPMI 1640 powder with phenol red and without HEPES and sodium bicarbonate (Life Technologies), 2.2 g/l sodium bicarbonate, 0.5 % (wt/vol) Albumax I (Life Technologies), 0.005 % (wt/vol) hypoxanthine, 0.03 % (wt/vol) l-glutamate, 25 μg/ml gentamicin. Dissolve hypoxanthine fully in a small volume of 1 M NaOH prior to addition; for 1 l of media, use 500 μl of NaOH to dissolve the required amount of hypoxanthine. Adjust the pH to 7.4 with NaOH or HCl. Sterilize media by filtration through a 0.22 μm membrane into a sterile container. Media may be stored at 4 °C for up to a month or until the pH changes (color of media becomes pink), whichever is sooner.
2.
Red blood cells: These are stored at 4 °C for a period no longer than a month from the time of blood drawing.
3.
Hypoxic gas: 3 % CO2, 4 % O2, 93 % N2.
2.2 Giemsa Smear
1.
Glass slides, grease-free.
2.
100 % methanol.
3.
Giemsa stain: This is diluted to 15 % in tap water just prior to use; do not prepare large volumes of diluted stain for storage.
2.3 Treatment and Staining of Parasites
1.
Sterile 96-well culture plates with lids, flat-bottom.
2.
Hypoxic culture chamber.
3.
Hoechst 33342: If purchased as a powder, dissolve in deionized water to obtain a 200 μg/ml stock solution. Sterilize with a 0.22 μm syringe filter and store at 4 °C in the dark.
4.
Fluo-4, AM (Life Technologies). Dilute to 5 mM in DMSO prior to use.
5.
Test compounds. If dissolved in DMSO, stock concentration should be at least 1000 times that of the screening concentration.
2.4 Imaging Flow Cytometry Data Acquisition and Analysis
1.
Phosphate-buffered saline (PBS) for sheath fluid.
2.
Amnis ImageStream system and INSPIRE software.
3.
IDEAS analysis software.
3 Methods
3.1 Treatment of Parasites
Parasitemia of the culture used for the assay is relatively high to allow for the rapid acquisition of events during flow cytometry. Culture and synchronization techniques are described in detail elsewhere [12, 13] and will not be extensively discussed here. All steps are performed in an appropriate biological safety cabinet with proper aseptic techniques.
2.
Perform a thin Giemsa smear [13] to assess parasitemia and parasite stage. Parasites should be roughly 32 h post-invasion (hpi) at the start of the experiment (Fig. 2) (see Note 2 ). Transfer culture to a conical centrifuge tube and pellet cells by centrifugation at 800 × g for 5 min. Remove culture medium and resuspend in fresh medium to 2.5 % hematocrit. Excessive parasitemia can be adjusted to 10 % by the addition of fresh erythrocytes.
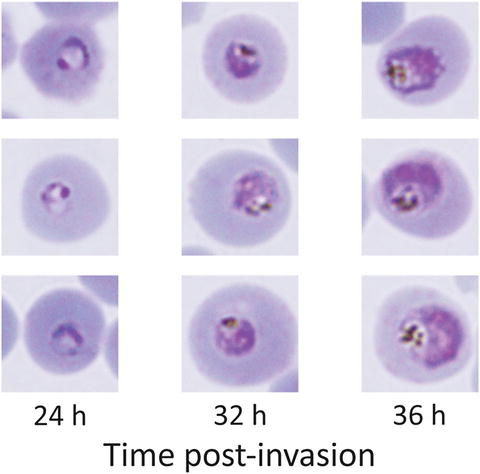
Fig. 2
Thin Giemsa smear of parasite culture. At the start of incubation with the test compounds, parasite stages should centre around 32 hpi, at the early to mid trophozoite stage. The parasite should occupy roughly a quarter of the area of the host erythrocyte, with prominent hemozoin pigment. Cultures with roughly 30 % of the parasites at the 24 or 36 hpi stages are considered well synchronized for this experiment; it is not necessary to obtain a very small window of synchrony
3.
Prepare working concentrations of the test compounds by dilution in PBS. This concentration should be 10 times that of the test concentration, i.e., the concentration at which the screen is performed (typically 10 μM for the preliminary screen; a working concentration of 100 μM is prepared for this screen). If compounds were originally dissolved in dimethyl sulfoxide (DMSO), the final concentration of DMSO should not exceed 0.1 % v/v. For the positive control, prepare chloroquine at a working concentration of 30 μM by dissolving in PBS. Avoid exposing all compounds to light unless the compounds are known to be photostable.
4.
In a sterile 96-well flat-bottomed microtiter plate, add 20 μl of the working concentration of each compound. Include a negative control (20 μl of PBS) and a positive control (20 μl of 30 μM chloroquine), as well as three additional wells for fluorescence compensation controls (20 μl of PBS each). Add 180 μl of the culture prepared in step 2 to each well and mix slowly to avoid generating bubbles (see Note 3 )
3.2 Staining of Parasites
1.
Prepare a sufficient amount of staining solution by diluting both Fluo-4 AM to 1 μM and Hoechst 33342 to 1 μg/ml together in prewarmed culture medium. Also prepare 300 μl solutions of each stain individually in culture medium at the same concentrations; these are single-stain controls for fluorescence compensation.
2.
Centrifuge microtiter plate at 800 × g for 1 min to pellet cells. Carefully aspirate supernatant so as to avoid disturbing the cell pellet. Wash cells by resuspending twice in 200 μl of prewarmed (37 °C) culture medium.
3.
Pellet cells by centrifugation at 800 × g for 1 min. Resuspend cells in 200 μl of staining solution. For the three wells reserved for fluorescence compensation, resuspend the cell pellet in (a) culture media without stain, (b) culture media with Fluo-4 AM, and (c) culture media with Hoechst 33342.
4.
Incubate the microtiter plate at 37 °C for 30 min.
5.
Wash cells twice in 200 μl of PBS. Concentrate cells by resuspending in 100 μl of PBS. This is roughly 5 % hematocrit, barring cell loss from the washing steps. Proceed immediately to ImageStream data acquisition (see Note 6 ).
3.3 ImageStream Acquisition
1.
Start the AMNIS ImageStream and the INSPIRE software as per the manufacturer’s instructions, including the loading of SpeedBeads and automated calibration. The sheath fluid used should be PBS.
2.
Acquire control samples for fluorescence compensation. Load the unstained samples. Use the Run/Setup function to make any minor adjustments such as the Core Tracking setting to centre cells in the field of view. Turn the bright field off by toggling the bright field on/off button in the Setup tab. Acquire at least 500 images of first the unstained cells, then the Fluo-4 single-stained cells, then the Hoechst 33342 single-stained cells for fluorescence compensation.
3.
 Quantitative Functional Morphology by Imaging Flow Cytometry
Principles of Amnis Imaging Flow Cytometry
Quantitative Functional Morphology by Imaging Flow Cytometry
Principles of Amnis Imaging Flow Cytometry
 Detection and Characterization of Rare Circulating Endothelial Cells by Imaging Flow Cytometry
Detection and Characterization of Rare Circulating Endothelial Cells by Imaging Flow Cytometry
 Assessment of Granulocyte Subset Activation: New Information from Image-Based Flow Cytometry
Assessment of Granulocyte Subset Activation: New Information from Image-Based Flow Cytometry
 Accurate Assessment of Cell Death by Imaging Flow Cytometry
Accurate Assessment of Cell Death by Imaging Flow Cytometry
 Quantitation of Chromosome Damage by Imaging Flow Cytometry
Quantitation of Chromosome Damage by Imaging Flow Cytometry




Turn the bright field back on. Choose a channel for the bright field apart from those used for the detection of Fluo-4 and Hoechst 33342 fluorescence (typically channels 3 and 2 respectively). Set the cell classifier to select for events that have an area between 50 and 500 in the bright-field channel (“Object Area” lower limit and upper limit for the bright-field channel), and mean fluorescent intensity between 50 and 500 in the Fluo-4 channel (“Mean Intensity” lower limit and upper limit for channel 3). Acquire at least 10,000 cells per test sample (see Note 7 ).
Related posts:
Quantitative Functional Morphology by Imaging Flow Cytometry
Principles of Amnis Imaging Flow Cytometry
Detection and Characterization of Rare Circulating Endothelial Cells by Imaging Flow Cytometry
Assessment of Granulocyte Subset Activation: New Information from Image-Based Flow Cytometry
Accurate Assessment of Cell Death by Imaging Flow Cytometry
Quantitation of Chromosome Damage by Imaging Flow Cytometry
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
