SECTION X PEDIATRIC IMAGING
Essentials 1
Case
A 6-month-old boy presents with abnormal head shape.

Questions
1. Which ONE of the following is NOT a secondary cause of these findings?
A. Shunted hydrocephalus
B. Rickets
C. Sickle cell anemia
D. Microcephaly
E. Prenatal polyhydramnios
2. Which disorder is often confused with asymmetric posterior positional plagiocephaly?
A. Unilateral lambdoid synostosis
B. Bilateral coronal synostosis
C. Sagittal synostosis
D. Metopic synostosis
E. Multiple suture synostosis
3. Which ONE of the following disorders is NOT commonly associated with craniosynostosis?
A. Apert syndrome
B. Crouzon syndrome
C. Neurofibromatosis type 1
D. Thanatophoric dysplasia
Answers and Explanations
Question 1
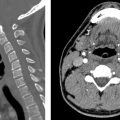
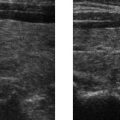
E. Correct! This is an example of craniosynostosis, premature fusion of the cranial sutures, as demonstrated by the surface-rendered computed tomography. Screening ultrasound image also demonstrates premature bony fusion represented by the solid hyperechoic calvarium in the expected location of the posterior sagittal suture. External compressive prenatal forces (prenatal constraint) may be a cause. Thus, oligohydramnios is a risk factor for premature closure of the cranial sutures, not polyhydramnios.
Other choices and discussion
A. Shunted hydrocephalus can cause secondary craniosynostosis. Inadequate intracranial pressure/inadequate separating forces on the patent sutures can cause premature closure.
B. Metabolic imbalance can cause secondary craniosynostosis. The exact mechanism is not well understood, but the mineral deficiencies in rickets delay vascularization of the growth plates and result in disorganization of chondrocytes and accumulation of osteoid at the metaphysis. Similarly, other calcium metabolic disorders are linked to premature cranial suture closure, including hypophosphatemia, vitamin D deficiency, renal osteodystrophy, and hypercalcemia.
C. Hematologic diseases that cause bone marrow hyperplasia are associated with premature cranial suture closure.
D. Microcephaly can cause secondary craniosynostosis. Similar to shunted hydrocephalus, inadequate intracranial pressure/inadequate separating forces on the patent sutures can cause premature closure.
Question 2
A. Correct! Unilateral lambdoid synostosis causes posterior plagiocephalic head shape, which is similar to the positional molding caused by infants who spend the majority of laying time on their backs. This latter phenomenon is also known as “flat head syndrome.” The incidence is estimated to be 1 in 100 infants, but the true incidence may be even greater to due campaigns to prevent sudden infant death syndrome such as “back to sleep” and “face up to wake up.” The difference between posterior plagiocephaly caused by positional molding and true unilateral lambdoid synostosis is crucial, as the former is treated conservatively with helmet therapy, while the latter is corrected surgically.
Other choices and discussion
B. Bilateral coronal synostosis causes brachycephaly. This head shape is characterized by a shortened anteroposterior dimension (“brachy” is the Greek root meaning short), resulting in a towering head shape.
C. Sagittal synostosis causes scaphocephaly. This head shape is characterized by an elongated anteroposterior dimension resulting in a boat-shaped head (“scapho” is the Greek root meaning boat). This head shape is also known as dolichocephaly.
D. Metopic synostosis causes trigonocephaly.
E. Severe multisuture synostosis causes a cloverleaf-shaped head. This is associated with multiple disorders.
Question 3
C. Correct! Neurofibromatosis type 1 can cause an abnormal head shape, but this is secondary to sphenoid dysplasia rather than craniosynostosis. In fact, sphenoid dysplasia is one of the many diagnostic criteria for neurofibromatosis type 1.
Other choices and discussion
A. Apert syndrome is an autosomal dominant disorder that is characterized by bilateral coronal suture craniosynostosis, midface hypoplasia, hypertelorism, and syndactyly. It is due to mutations in the fibroblast growth factor receptor 2 gene.
B. Crouzon syndrome is also an autosomal dominant disorder caused by mutations in the fibroblast growth factor receptor 2 gene. However, this syndrome is typically characterized by multiple sutural synostoses. When severe, it manifests as a cloverleaf skull. Syndactyly is not typical in Crouzon syndrome.
D. Type 2 thanatophoric dysplasia is typically caused by a new, sporadic, dominant mutation in the fibroblast growth factor receptor 2 gene. The typical skull manifestation results in a cloverleaf skull due to severe multiple suture craniosynostosis.
Suggested Readings
Dover MS. Abnormal skull shape: clinical management. Pediatr Radiol 2008;38(Suppl 3):S484–S487 Peitsch WK, Keefer CH, LaBrie RA, Mulliken JB. Incidence of cranial asymmetry in healthy newborns. Pediatrics 2002;110:e72 Sze RW, Parisi MT, Sidhu M, et al. Ultrasound screening of the lambdoid suture in the child with posterior plagiocephaly. Pediatr Radiol 2003;33:630–636Top Tips
Multidetector computed tomography with three-dimensional reformations is the gold standard for imaging diagnosis of craniosynostosis.
Targeted high-frequency ultrasound is an effective screening tool.
Craniosynostosis can be primary or secondary. Secondary causes include microcephaly, overshunted hydrocephalus, hematologic conditions that cause marrow hyperplasia, and calcium metabolic disorders such as rickets.
Essentials 2
Case

Questions
1. Why does this type of pneumonia occur more often in children than in adults?
A. Different bacteria are present in the two populations.
B. The collateral airways are underdeveloped in children.
C. The immune system in children organizes the infection differently than in adults.
D. Clinical symptoms in children present earlier than in adults, resulting in the detection of the atypical rounded appearance more often.
2. Which of the follow features can help differentiate other disease processes from the disorder featured in the test case? (Select ALL that are correct.)
A. Multiplicity
B. Air-fluid level
C. Calcification
D. Bony erosion of underlying ribs
3. What is the most common pulmonary metastatic lesion in children?
A. Wilms tumor
B. Neuroblastoma
C. Osteosarcoma
D. Ewing sarcoma
E. Medulloblastoma
Answers and Explanations
Question 1
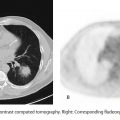
B. Correct! This is rounded pneumonia (PNA). The collateral pathways of air circulation (channels of Lambert and pores of Kohn) are incompletely developed in children under the age of 8 years. As bronchopneumonia infection attempts to spread, the lack of collateral pathways creates a rounded appearance.
Other choices and discussion
A. Most commonly, Streptococcus pneumoniae is responsible for rounded PNA in children. This pathogen is also common in adult community-acquired PNA.
C. It is not the immune system, but rather the anatomic variance, that creates rounded PNA.
D. The timing of symptoms is similar in adults and children, and this is not responsible for rounded PNA.
Question 2
A. Correct! Round PNA is typically solitaire. Multiple lesions are more likely to result from other infections (such as fungal) or metastasis.
B. Correct! Air-fluid levels are unusual in round PNA, but can be seen with lung abscess, necrotizing PNA, or an infected bronchogenic cyst.
C. Correct! Calcifications are not present in round PNA, but can be present in thoracic neuroblastoma, fungal infection, pulmonary carcinoid, hamartomas, or osteosarcoma metastasis.
D. Correct! When a thoracic mass is seen in conjunction with rib erosion or soft tissue spread, neuroblastoma should be suspected. Bony involvement is not present in pneumonia.
Question 3
A. Correct! Wilms tumor is the most common primary neoplasm to spread to the lungs. Note that metastatic pulmonary masses are much more common than primary pulmonary malignancies in children.
Other choices and discussion
B. Neuroblastoma pulmonary metastases are rare, and when present, portend a poor prognosis. More commonly, neuroblastoma metastases occur in the bone.
C. Osteosarcoma pulmonary metastases are the second most common cause of pediatric pulmonary metastases, after Wilms tumor. This metastatic lesion can also present with a spontaneous pneumothorax.
D. Although the lung is the most common site of the metastasis for Ewing sarcoma and for many other sarcomas in childhood, it is not the most common cause of metastasis to the lungs.
E. The most frequent of spread of metastasis for medulloblastoma is through the cerebrospinal fluid. Lung metastasis is uncommon.
Suggested Readings
Dishop MK, Kuruvilla S. Primary and metastatic lung tumors in the pediatric population: a review and 25-year experience at a large children’s hospital. Arch Pathol Laboratory Med 2008;132:1079–1103 Kim Y-W, Donnelly LF. Round pneumonia: imaging findings in a large series of children. Pediatric Radiology 2007;37:1235–1240Top Tips
Round PNA is seen in children < 8 years of age due to underdevelopment of collateral air pathways. If the typical clinical presentation of PNA is present, then no further imaging is required at the time of diagnosis.
Follow up imaging can be considered after an appropriate course of antibiotics, especially if symptoms do not improve.
Metastases are much more common than primary tumors of the lung in children. Wilms tumor is the most common pediatric pulmonary metastatic lesion.
Essentials 3
Case
An incidental finding in an asymptomatic pediatric patient.

Questions
1. Which ONE of the following imaging features is most characteristic of this bone finding?
A. Ground glass appearance of the matrix
B. Involvement of multiple bones
C. Cortical destruction
D. T2 hyperintensity
E. Ring and arc appearance of the matrix
2. Which ONE of the following entities is characterized by polyostotic fibrous dysplasia, endocrine dysfunction, and cutaneous hyperpigmentation?
A. Jaffe-Campanacci syndrome
B. Cherubism
C. Mazabraud syndrome
D. McCune-Albright syndrome
3. Although early Caffey disease can be confused with fibrous dysplasia, cortical involvement and periostitis are typically not seen in fibrous dysplasia. Which ONE of the following is a feature of Caffey disease?
A. Most common site of involvement is the mandible
B. Patients are asymptomatic
C. Occurs in children > 6 months of age
D. Epiphyses are involved
E. Manifestations usually persist past 2 years of age
Answers and Explanations
Question 1
A. Correct! This is fibrous dysplasia (FD). A ground glass matrix with expansion of the marrow space is the most characteristic finding in FD. Computed tomography is the preferred modality to evaluate suspected cases of FD.
Other choices and discussion
B. Although FD can be polyostotic, this is not a defining characteristic. In fact, the majority of cases of FD are monostotic.
C. Cortical destruction is an aggressive feature and should not be present in FD (which is a benign nonaggressive entity).
D. The fibrous component of FD causes T2 hypointensity, not hyperintensity.
E. A ring and arc internal matrix is characteristic of cartilaginous lesions, not FD.
Question 2
D. Correct! These features best describe McCune-Albright syndrome. The endocrine dysfunction is typically precocious puberty, and the hyperpigmentation results in café au lait spots.
Other choices and discussion
A. Jaffe-Campanacci syndrome is characterized by multiple nonossifying fibromas, not by FD. Other features of this entity include café au lait spots and hypogonadism.
B. Cherubism is defined by autosomal dominant FD of the bilateral jaw.
C. Mazabraud syndrome consists of polyostotic FD and intramuscular myxomas.
Question 3
A. Correct! The mandible is the most common location for Caffey disease.
Other choices and discussion
B. Patients with Caffey disease are typically symptomatic. There is a classic triad of fevers, irritability, and soft tissue swelling.
C. Although there have been case reports of Caffey disease in older children, it typically occurs in children < 6 months of age.
D. The cortical hyperostosis spares the epiphysis in Caffey disease.
E. Caffey disease symptoms usually resolve 6 to 12 months after initial presentation. There are rare cases of persistent or protracted courses, but these are atypical.
Suggested Readings
Belsuzarri TAB, Araujo JFM, Melro CAM, et al. McCune-Albright syndrome with craniofacial dysplasia: clinical review and surgical management. Surg Neurol Int 2016;7(Suppl 6):S165–S169 Shandilya R, Gadre KS, Sharma J, Joshi P. Infantile cortical hyperostosis (caffey disease): a case report and review of the literature—where are we after 70 years? J Oral Maxillofacial Surg 2013;71:1195–1201Top Tips
A ground glass appearance on computed tomography is most characteristic of FD.
McCune-Albright syndrome consists of polyostotic FD, precocious puberty, and café au lait spots.
Caffey disease occurs in symptomatic children who are < 6 months of age, commonly involves the mandible, and is manifested radiographically by periosteal reaction that spares the epiphysis.
Essentials 4
Case
A patient presents with a history of hip dysplasia.

Questions
1. What are the normal alpha angle and femoral head coverage?
A. Greater than 60 degrees and 40%
B. Greater than 60 degrees and 50%
C. Greater than 30 degrees and 40%
D. Greater than 30 degrees and 50%
2. Which ONE of the following is a risk factor for hip dysplasia?
A. Cephalic presentation
B. Family history of hip dysplasia
C. Polyhydramnios
D. Male sex
E. Arm deformity
3. What are the criteria for functional immaturity of the hips?
A. Age < 12 weeks, alpha angle 50 to 60 degrees, and femoral head coverage of 40 to 50%
B. Age < 12 weeks, alpha angle 40 to 60 degrees, and femoral head coverage of 40 to 50%
C. Age < 16 weeks, alpha angle 50 to 60 degrees, and femoral head coverage of 35 to 50%
D. Age < 16 weeks, alpha angle 40 to 60 degrees, and femoral head coverage of 35 to 50%
Answers and Explanations
Question 1
B. Correct! An alpha angle > 60 degrees and femoral head coverage of > 50% are normal for patients who are > 12 weeks old. On radiographs, the acetabular angle is the complementary angle to the alpha angle on ultrasound, and thus the normal acetabular angle should be < 30 degrees. This patient has developmental dysplasia of the hip.
Question 2
B. Correct! Family history is a risk factor for developmental dysplasia of the hips.
Other choices and discussion
A. Breech presentation is a risk factor for developmental dysplasia of the hips.
C. Oligohydramnios is a risk factor for developmental dysplasia of the hips.
D. Female sex is a risk factor for developmental dysplasia of the hips and thought to be due to increased sensitivity to the maternal hormone relaxin.
E. Foot deformity, not arm deformity, is a risk factor for developmental dysplasia of the hips.
Question 3
A. Correct! In children < 12 weeks old, laxity of the hip is allowable. This phenomenon is called functional immaturity of the hips and is secondary to maternal estrogen influence and increased ligamentous laxity. In these cases, the alpha angles are between 50 to 60 degrees and the femoral head coverage between 40 and 50%. Up to 90% of cases identified by ultrasound will normalize upon follow up. Thus, a follow up examination is suggested in 4 to 6 weeks.
Suggested Readings
Roof AC, Jinguji TM, White KK. Musculoskeletal screening: developmental dysplasia of the hip. Pediatric Annals 2013;42: e238–e244 US Preventive Services Task Force. Screening for developmental dysplasia of the hip: recommendation statement. Pediatrics 2006;117:898–902Top Tips
The normal alpha angle of the hip on ultrasound is > 60 degrees, and the normal femoral head coverage is > 50%.
Children who are < 12 weeks old may have increased laxity of the hips and can normally have a slightly lower alpha angle and femoral head coverage (50 degrees and 40% coverage). This is called functional immaturity of the hips.
Ultrasound is the preferred modality for evaluating the hip before the age of 6 months. Once there is significant ossification of the proximal femoral epiphyses, then radiographs are required.
Essentials 5
Case
A patient presents with a history of acute unilateral pelvic pain.
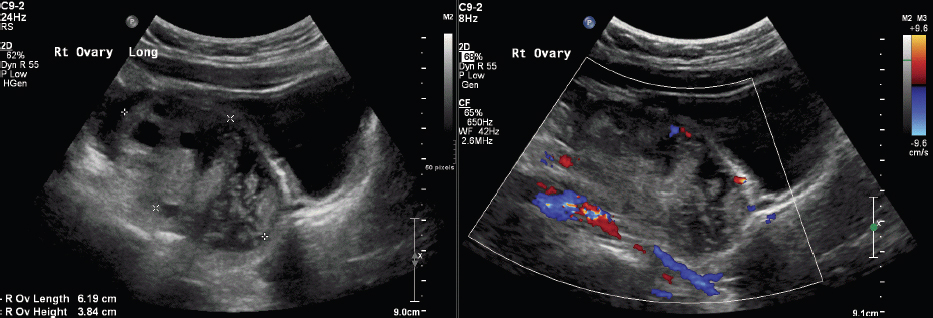
Questions
1. Which ONE of the following is the BEST predictor of ovarian torsion?
A. Lack of arterial Doppler waveforms
B. Unilateral enlarged ovarian size and volume
C. Medialization of the ovary
D. Peripherally displaced follicles
E. Presence of an ovarian mass
2. Which ONE of the following does NOT predispose female pediatric patients to the disorder presented in this test case?
A. Ovarian mass
B. Ovarian cyst
C. Maternal hormonal stimulation
D. Hydrosalpinx
E. Large body habitus
3. Which ONE of the following is the BEST predictor of testicular torsion?
A. Lack of intraparenchymal vascular flow
B. Unilateral enlarged testicular size and volume
C. Medialization of the testicle
D. Microlithiasis
E. Presence of an intratesticular mass
Answers and Explanations
Question 1
B. Correct! A unilaterally enlarged ovary is the best indicator of torsion. Asymmetry correlating to the clinical side of symptoms is also an important indicator of torsion. Some studies have described an ovarian length > 5 cm or a volume > 20 mL as sensitive markers of ovarian torsion in children.
Other choices and discussion
A. Although at first glance the absence of arterial waveforms might seem like the most intuitive answer, Doppler findings in cases of ovarian torsion are highly variable, ranging from normal venous and arterial waveforms to completely absent or reversed waveforms. This is a result of the dual blood supply of the ovaries (uterine and ovarian arteries), timing of presentation, and potential intermittent nature of ovarian torsion. Therefore, the presence of intraparenchymal vascular flow (venous or arterial) does not exclude ovarian torsion.
C. Medialization of an ovary is an important secondary sign of torsion, but it is not as sensitive as ovarian size. Medialization refers to twisting of the vascular pedicle, which secondarily displaces the ovary toward the uterus.
D. Peripheral displacement of ovarian follicles is an important secondary sign of torsion, but it is not as sensitive as ovarian size. This finding is due to parenchymal edema/vascular engorgement, which displaces the follicles outward.
E. An ovarian mass predisposes the vascular pedicle to twist, as the mass can act as a fulcrum for the torsion. However, ovarian torsion does not require the presence of a mass, and even when present, concurrent masses are not as sensitive for the detection of ovarian torsion as unilateral ovarian enlargement.
Question 2
E. Correct! There is no correlation between a large body habitus and ovarian torsion.
Other choices and discussion
A. An ovarian solid mass can act as a fulcrum for torsion.
B. An ovarian cyst can act as a fulcrum for torsion.
C. Hormonal stimulation causes ovarian enlargement, which can predispose the ovary to torsion. This is one cause of fetal ovarian torsion.
D. Hydrosalpinx is associated with up to 9% of cases of ovarian torsion.
Question 3
A. Correct! In contrast to ovarian torsion, the most sensitive sign of testicular torsion is lack of parenchymal flow. This is due to a single source of vascular supply. Intermittent testicular torsion should be suspected when there is initial lack of parenchymal flow with subsequent positive parenchymal flow during an ultrasound examination. Evaluation of the spermatic cord is also important, as swirling of the cord or vascular engorgement are sensitive signs for intermittent testicular torsion as well.
Other choices and discussion
B. Although an enlarged testicle is often seen in torsion, it is not as sensitive as lack of intraparechymal flow. In addition, an enlarged testicle can also be associated with orchitis and testicular neoplasia.
C. Change in the position of the testicle has not been reported as a sign of testicular torsion.
D. Microlithiasis has not been shown to be predictive of testicular torsion. However, there is an association of microlithiasis with germ cell tumor. If microlithiasis is isolated, then no further imaging workup is required. Follow up should be suggested only in patients with an increased risk of germ cell tumor, including personal or family history, maldescent or undescended testes, orchiopexy, or testicular atrophy.
E. Presence of an intratesticular mass does increase the risk of testicular torsion. An abnormally high attachment of the tunica vaginalis results in a bell clapper deformity, which allows the testicle to freely rotate in the scrotum and predisposes to testicular torsion.
Suggested Readings
Munden MM, Williams JL, Zhang W, et al. Intermittent testicular torsion in the pediatric patient: sonographic indicators of a difficult diagnosis. Am J Roentgenol 2013;201:912–918 Ngo A-V, Otjen JP, Parisi MT, et al. Pediatric ovarian torsion: a pictorial review. Ped Radiol 2015;45:1845–1855 Richenberg J, Belfield J, Ramchandani P, et al. Testicular microlithiasis imaging and follow-up: guidelines of the ESUR scrotal imaging subcommittee. Eur Radiol 2015;25:323–330 Ringdahl E, Teague L. Testicular torsion. Am Fam Physician 2006;74:1739–1743 Waldert M, Klatte T, Schmidbauer J, et al. Color doppler sonography reliably identifies testicular torsion in boys. Urology 2010;75:1170–1174Top Tips
An enlarged ovary in the setting of acute unilateral pediatric pelvic pain represents ovarian torsion until proven otherwise.
Absence of ovarian vascular flow is unreliable in the diagnosis of ovarian torsion. This is partially due to a dual blood supply from the ovarian and uterine vessels.
In contrast to ovarian torsion, lack of vascular flow is the most reliable sign of testicular torsion. Detection prior to 6 hours has a good salvage rate.
Essentials 6
Case

Questions
1. Which ONE of the following is characteristic of this entity?
A. Large posterior fossa
B. Flattening of the tectum
C. Tonsillar herniation
D. Small massa intermedia
E. Vermian hypoplasia
2. Which ONE of the following prenatal signs does NOT suggest this entity?
A. Lemon sign
B. Strawberry sign
C. Myelomeningocele
D. Elevated alpha-fetoprotein
E. Banana sign
3. Which ONE of the following is NOT associated with syringomyelia?
A. Chiari 1 malformation
B. Chiari 2 malformation
C. Scoliosis
D. Klippel-Feil syndrome
E. Leukemic involvement of the spine
Answers and Explanations
Question 1
C. Correct! This image is characteristic of Chiari malformation type 2, with inferior cerebellar tonsillar herniation, beaked tectum, small posterior fossa, and large massa intermedia. Tonsillar herniation is the most characteristic feature of Chiari malformations. Inferior cerebellar tonsillar herniation > 6 mm below the foramen magnum in symptomatic patients is considered abnormal.
Other choices and discussion
A. The features of Chiari malformation type 2 include a small (not a large) crowded posterior fossa. A large posterior fossa can be seen in Dandy Walker malformations and other associated entities.
B. In Chiari 2, the configuration of the tectum is often beaked, not flattened. This represents fusion of the midbrain colliculi, which are directed posteriorly and invaginate into the cerebellum.
D. A large massa intermedia, which is an interthalamic adhesion (situated above the tectum and midline), is characteristic of Chiari 2 malformation.
E. Vermian hypoplasia is most commonly associated with Dandy Walker malformations. Herniation of the vermis is seen in Chiari 2 malformations, but the vermis is not hypoplastic in these cases.
Question 2
B. Correct! The “strawberry” sign is most commonly associated with trisomy 18. The strawberry sign refers to a flattened occiput and a pointed anterior calvarium. These are associated with hypoplasia of the occipital and frontal bones, causing brachycephaly.
Other choices and discussion
A. The “lemon” sign refers to a biconcave appearance of the frontal bones due to the open neural defect of the spine associated with Chiari 2 malformation. It is rarely seen in fetuses > 24 weeks, secondary to decreased pliability of the calvarium and increased hydrocephalus.
C. By definition, Chiari 2 malformation contains an open neural tube defect, most commonly a myelomeningocele.
D. Elevated maternal alpha-fetoprotein is best evaluated at 15 to 20 weeks of gestation. When elevated, it is suggestive of an open neural tube defect (a prerequisite component of Chiari 2, as discussed above) and should be correlated with the 20-week prenatal ultrasound screening examination. In addition to open neural tube defects, an elevated alpha-fetoprotein is also associated with ventral abdominal wall defects.
E. On axial imaging of the fetal posterior fossa, the “banana” sign represents crowding of the posterior fossa. Specifically, the cerebellar hemispheres form a banana shape as they wrap around the brainstem. This is due to downward herniation of those elements secondary to a tethered cord and an open neural tube defect.
Question 3
E. Correct! Leukemic involvement of the spine affects the bone marrow but not the cord itself.
Other choices and discussion
A and B. Syringomyelia (as known as syrinx) is a cystic spinal cord cavity that is not contiguous with the central cord canal and is due to alterations of cerebrospinal fluid (CSF) flow. Both Chiari 1 and 2 malformations can cause syringomyelia. A screening magnetic resonance imaging examination of the spine should be performed to rule out a syrinx at initial diagnosis of a Chiari malformation.
C. Scoliosis is not a cause for syringomyelia. However, many studies suggest that scoliosis develops in the presence of a syrinx. It is postulated that abnormal intramedullary pressure in the spinal cord causes interference with the postural tonic reflexes. Up to 82% of children with a syrinx will have scoliosis.
D. Klippel-Feil syndrome can cause alterations of CSF flow similar to that of severe scoliosis. In addition, skull and upper cervical spine fusion anomalies associated with the syndrome can also alter CSF flow, mimicking the dynamics of a Chiari malformation.
Suggested Readings
Atalar MH, Salk I, Egilmez H. Classical signs and appearances in pediatric neuroradiology: a pictorial review. Polish J Radiol 2014;79:479–489 Driscoll DA, Gross SJ. Screening for fetal aneuploidy and neural tube defects. Genetics in Med 2009;11:818–821 Eule JM, Erickson MA, O’Brien MF, et al. Chiari I malformation associated with syringomyelia and scoliosis: a twenty-year review of surgical and nonsurgical treatment in a pediatric population. Spine 2002;27:1451–1455 Kagawa M, Jinnai T, Matsumoto Y, et al. Chiari I malformation accompanied by assimilation of the atlas, Klippel-Feil syndrome, and syringomyelia: case report. Surgical Neurol 2006;65:497–502Top Tips
Chiari 1 malformation is peg-like in configuration, and the cerebellar tonsillar herniation below the foramen magnum measures 6 mm or greater in symptomatic patients. A screening spinal imaging study should be performed to evaluate for the presence of a syrinx upon initial diagnosis of a Chiari 1 malformation.
Chiari 2 malformation is always associated with an open spinal defect, most commonly a lumbosacral myelomeningocele.
The “lemon” and “banana” signs represent prenatal sequelae of a Chiari 2 malformation.
Essentials 7
Case
A patient presents with a history of congenital pulmonary abnormality.

Questions
1. Which of the following features differentiate pure congenital pulmonary airway malformation from pulmonary sequestration?
A. Cystic lung disease
B. Most commonly occurs in the left upper lobe
C. Lack of systemic arterial supply
D. Solid component
E. None of the above
2. Which ONE of the following is NOT helpful in differentiating between intralobar and extralobar sequestration?
A. Venous drainage
B. Arterial supply
C. Typical age of presentation
D. Pleural investment
3. Which ONE of the following presents as a solid lung lesion?
A. Bronchial atresia
B. Congenital lobar overinflation
C. Diaphragmatic hernia
D. Type 3 congenital pulmonary airway malformation
E. Type 1 pleuropulmonary blastoma
Answers and Explanations
Question 1
C. Correct! The test case demonstrates a cystic lesion within the left upper lobe, without aberrant systemic arterial supply or internal lung markings, most consistent with a purely macrocystic congenital pulmonary airway malformation (CPAM). Pure CPAM lesions should not contain a systemic arterial supply, but instead should have a pulmonary artery supply. This is in contradistinction to pulmonary sequestrations that always have a systemic arterial supply and no airway connection. Hybrid CPAMs are lesions with airway connections and systemic arterial supply.
Other choices and discussion
A. Both CPAM and sequestrations can contain cystic components. Type 1 CPAMs are macrocystic, whereas type 2 CPAMs contain smaller cysts.
B. A distinct pattern of lobar involvement has not been described in CPAM. Pulmonary sequestration is typically seen in the lower lobes, with the left side more often involved than the right. The left upper lobe is most commonly affected in congenital lobar overinflation, followed by the right middle lobe, and then the right upper lobe.
D. Solid components can be present in both CPAMs and sequestrations.
Question 2
B. Correct! Arterial supply does not help differentiate between intra- and extralobar sequestration, as both have a systemic arterial supply.
Other choices and discussion
A. Intralobar sequestration typically has pulmonary venous drainage, whereas extralobar sequestration drains systemically.
C. Intralobar sequestration typically presents in later childhood with recurrent pneumonias in the same location. Extralobar sequestration typically presents in the prenatal/neonatal period.
D. Extralobar sequestration has a separate pleural investment, whereas intralobar sequestration shares the pleural investment with the adjacent normal lung. This difference is usually not evident radiographically.
Question 3
D. Correct! Microcystic CPAM (type 3) presents as a solid mass, not a lucent lesion, as is seen in type 1 and 2 lesions (macrocystic and small cysts, respectively).
Other choices and discussion
A. Interruption of a bronchus with hyperlucent lung distally is most characteristic of bronchial atresia. Oftentimes, mucous can fill the interrupted bronchus, creating a “finger in glove” appearance.
B. Congenital lobar hyperinflation is the end result of airway insult and is represented by dilated alveoli. When imaged early in life, these may present as opaque consolidations. However, the opaque appearance is due to incomplete clearance of fluid, and with serial imaging, there will be eventual clearance to a hyperlucent, air-filled lung.
C. Although fluid within herniated bowel can have a solid appearance, some gas is typically present. Thus, this choice is excluded. In fact, gas in herniated bowel can often mimic a lucent lung lesion.
E. Pleuropulmonary blastomas have variable appearances in the chest, including lucent lesions. They are categorized by intralesional components. Air-filled lucent lesions are type 1. Cystic lesions with variable solid components are type 2. Solid lesions with mass effect are type 3.
Suggested Readings
Biyyam DR, Chapman T, Ferguson MR, et al. Congenital lung abnormalities: embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics 2010;30:1721–1738Top Tips
Radiographically, there are three types of CPAMs: macrocystic, small cysts, and microcystic/solid.
Pulmonary sequestrations have an arterial systemic supply and lack communication to the airways.
Intralobar sequestrations have pulmonary venous drainage, whereas extralobar sequestrations have systemic venous drainage. Intralobar sequestrations often present with recurrent same-site pneumonia.
Essentials 8
Case
A patient presents with a history of trauma.

Questions
1. Which ONE of the following is consistent with nonaccidental trauma?
A. Metaphyseal fraying
B. Diffuse osteopenia
C. Widening of the physis
D. Metaphyseal corner fracture
2. The proposed mechanism for a classic metaphyseal lesion is which of the following?
A. Isolated avulsion fracture at the corner of the metaphysis
B. Compressive axial force along the axis of the metaphysis
C. Transmetaphyseal disruption of the trabeculae of the primary spongiosa due to shear or bending forces
D. Direct perpendicular force to the axis of the diaphysis creating propagation of the fracture through the metaphysis
3. After detection of a classic metaphyseal lesion, what is the next BEST step for the radiologist?
A. Urgent call to the referring physician, and request a skeletal survey.
B. Call to the on-call social worker.
C. Report the findings as usual, as most clinicians understand what a classic metaphyseal lesion signifies.
D. Call law enforcement.
Answers and Explanations
Question 1
D. Correct! This is case of nonaccidental trauma (NAT), with a skull fracture of the parietal bone, multiple healing bilateral posterior rib fractures, and a corner metaphyseal fracture of the distal tibia. Corner metaphyseal fractures are essentially diagnostic of NAT, while all of the remaining listed choices are characteristic of rickets instead. There is some minor controversy on whether healed rickets can have a similar appearance to the metaphyseal corner fracture, aka classic metaphyseal lesion (CML), but if the other listed features of rickets are absent, the CML remains highly suspicious for NAT.
Question 2
C. Correct! As stated above, CML is a transmetaphyseal fracture due to shear or bending forces through the primary spongiosa.
Other choices and discussion
A. Although CML is also known as a metaphyseal corner fracture, it represents more than simply an avulsion injury. The CML is also known as a bucket handle fracture, which more accurately describes the transmetaphyseal fracture course.
B. Compressive forces at the metaphysis are classically associated with a Salter-Harris V fracture of the physis. These fractures are rare.
D. Direct perpendicular force to the axis of the diaphysis can cause transverse fractures or fractures with a butterfly segment. If they do extend to the metaphysis, a CML should not be present.
Question 3
A. Correct! CMLs are strongly associated with child abuse, and in 95% of the cases, an additional traumatic injury is present. A skeletal survey is an effective way of identifying additional osseous injuries in this vulnerable population and has been shown to detect another fracture in up to 87% of the cases.
Other choices and discussion
B. Although a social work consultation is appropriate, this is not the primary responsibility of the radiologist. The urgent call to the referring clinician will prompt this social work evaluation.
C. As stated above, a CML is highly suspicious for NAT, and the responsibility of conveying the seriousness of this finding belongs to the radiologist.
D. Although law enforcement may need to become involved, this decision should be made by the clinicians and social workers after a complete evaluation is performed.
Suggested Readings
Thackeray JD, Wannemacher J, Adler BH, et al. The classic metaphyseal lesion and traumatic injury. Ped Radiol 2016;46(8):1128–1133 Wood BP. Commentary on “a critical review of the classic metaphyseal lesion: traumatic or metabolic?” Am J Roentgenol 2014;202:197–198Top Tips
CML, also known as a metaphyseal bucket handle or corner fracture, is highly suspicious for NAT.
When a CML is detected, direct communication should be made to the referring physician, and a skeletal survey should be performed.
Radiographic findings of rickets include diffuse osteopenia, fraying or cupping of the metaphysis, and widening of the physis.
Essentials 9
Case
A pediatric patient with a history of seizures.

Questions
1. Which ONE of the following is MOST characteristic of tuberous sclerosis complex?
A. Subcortical tubers
B. Band heterotopia
C. Cardiac lipomas
D. Bone cysts
E. Posterior fossa hemangioblastoma
2. Adenoma sebaceum (angiofibromas of the skin), subependymal nodules, bone islands, and cardiac rhabdomyomas associated with tuberous sclerosis complex are categorized as which of the following?
A. Malignant tumors
B. Hamartomas
C. Teratomas
D. Dysplasias
3. How can subependymal nodules best be differentiated from subependymal giant cell astrocytomas on imaging?
A. Size
B. Location
C. Calcification
D. Contrast enhancement
E. Magnetic resonance spectroscopy
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


