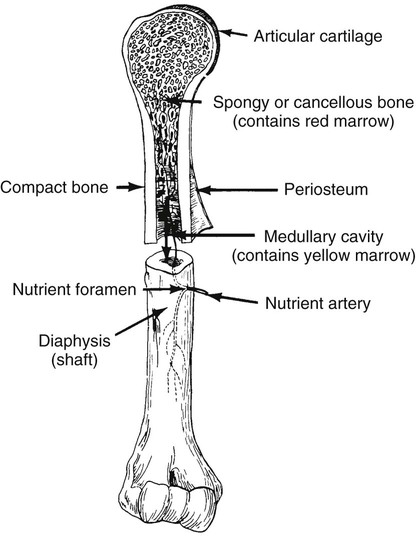
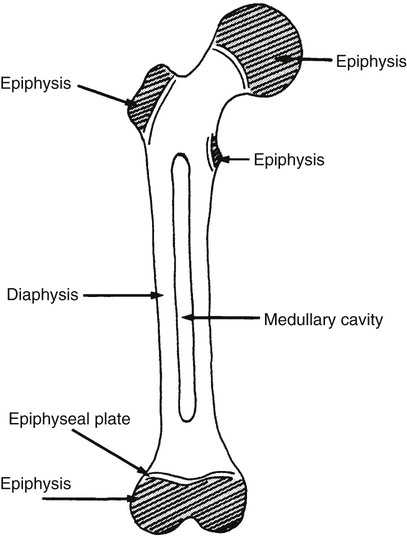
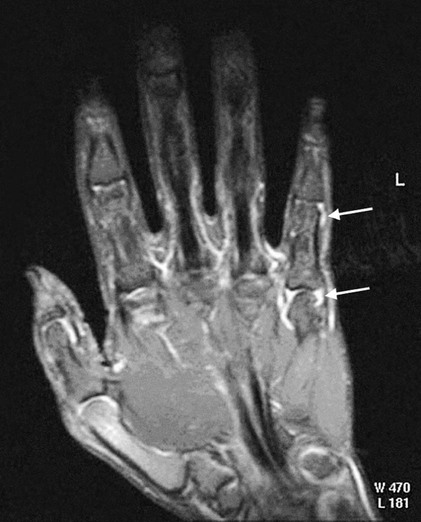
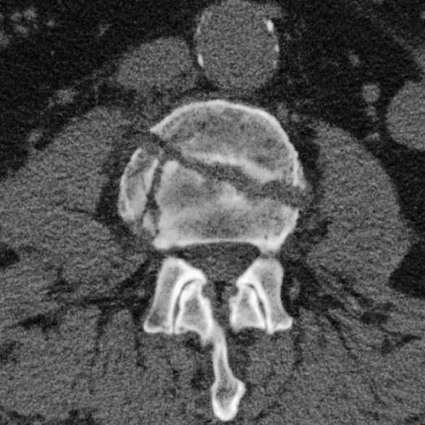
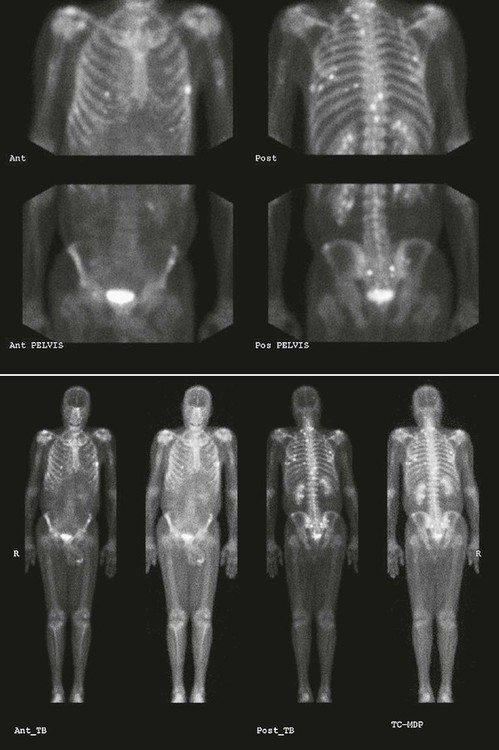
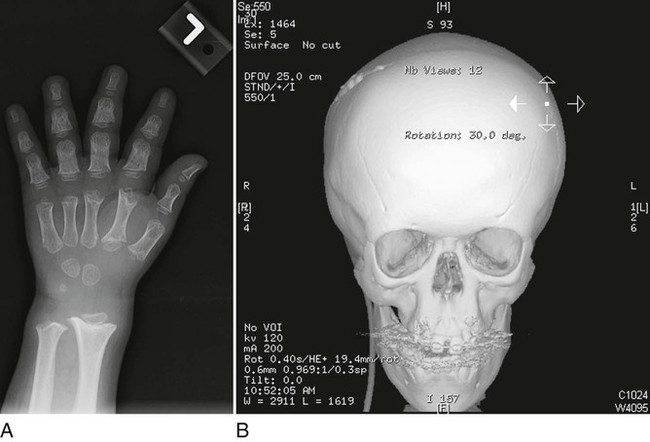
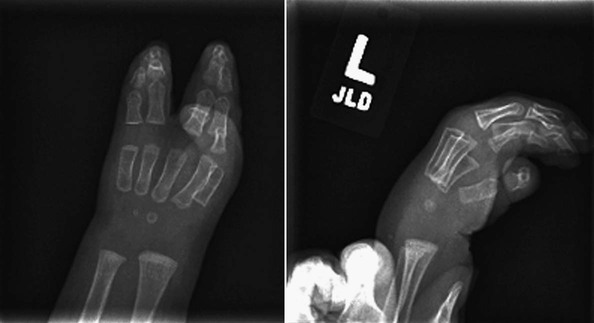
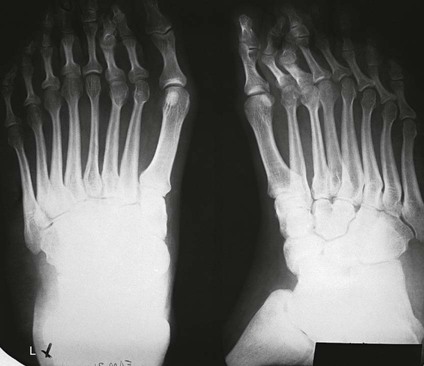
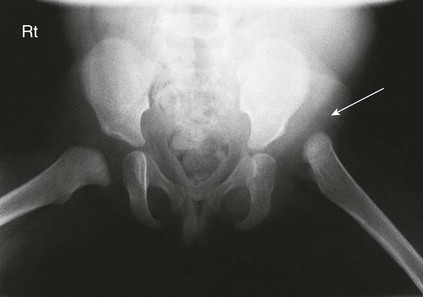
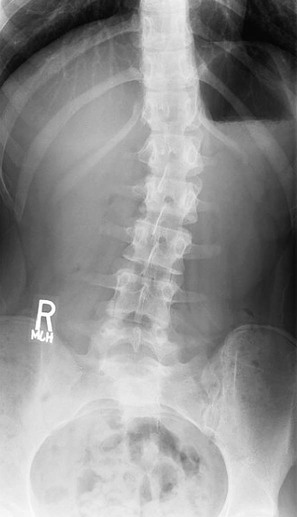
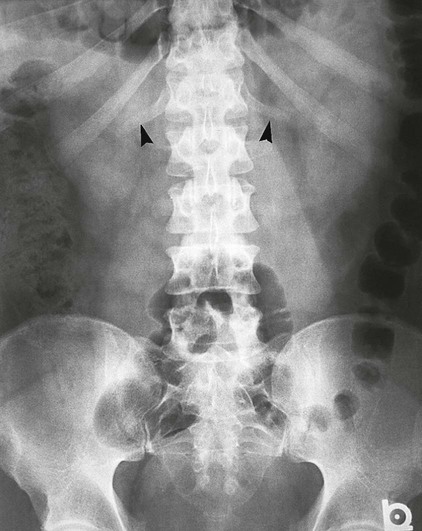
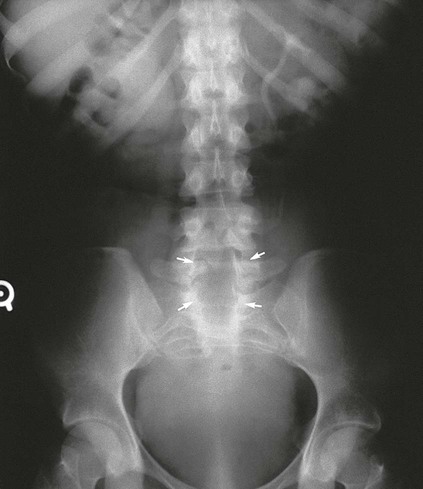
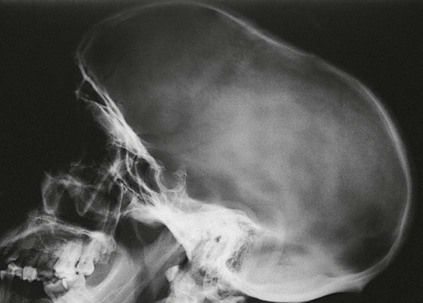
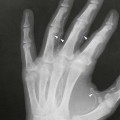
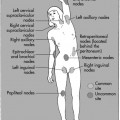
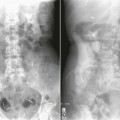
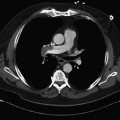
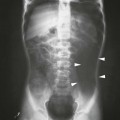
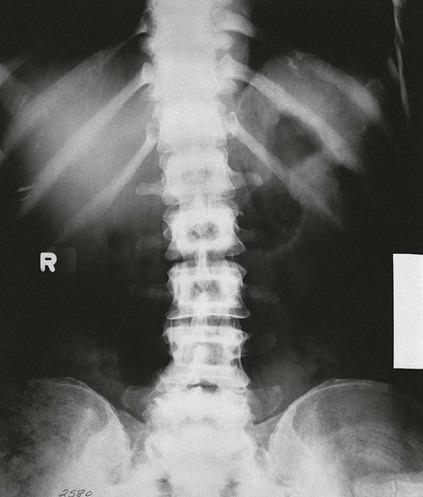
On completion of Chapter 2, the reader should be able to do the following: • Describe the anatomic components of the skeletal system on a macroscopic level and basic microscopic level. • Identify and explain the criteria for assessing technical adequacy of skeletal radiographs. • Characterize a given condition as congenital, inflammatory, arthritic, or neoplastic. • Specify the pathogenesis, signs and symptoms, and prognosis of the skeletal pathologies cited in this chapter. • Explain the role of various imaging modalities in the diagnosis and treatment of skeletal pathologies. The skeletal system is composed of 206 separate bones and is responsible for body support, protection, movement, and blood cell production. It contains more than 98% of the body’s total calcium and up to 75% of its total phosphorus. The system is commonly divided into the axial skeleton (Fig. 2-1), which contains 80 bones, and the appendicular skeleton (Fig. 2-2), which contains 126 bones. Bone is a type of connective tissue, but it differs from other connective tissue because of its matrix of calcium phosphate. The construction of this matrix further classifies bone tissue as either compact (dense) or cancellous (spongy) (Fig. 2-3). The bones of the skeletal system may also be classified according to their shape to include long, short, flat, and irregular bones. The diaphysis of a long bone refers to the shaft portion and is the primary site of ossification, whereas the epiphysis refers to the expanded end portion and is the secondary site of ossification (Fig. 2-4). The metaphysis refers to the growth zone between the epiphysis and the diaphysis. It is the area of greatest metabolic activity in a bone. A cartilaginous growth plate is located between the metaphysis and the epiphysis in the bone of a growing child. Radiographically, these growth areas appear radiolucent. As the body matures, this cartilage calcifies and is no longer radiographically visible in the adult. Magnetic resonance imaging (MRI) is an important modality used in imaging of skeletal pathology, particularly in providing soft tissue detail because of its superior contrast resolution. It is considered the modality of choice for detection and staging of soft tissue tumors involving the extremities. It is also extremely useful in evaluation of joints, particularly the knee and the shoulder (Fig. 2-5). Newer imaging techniques and improved equipment allow MRI to detect a greater number of musculoskeletal subtleties with higher-resolution imaging (Fig. 2-6). Sometimes these subtleties mimic bone pain but involve soft tissues instead, which is an important distinction. Bone marrow imaging done with MRI is superior to other modalities in visualizing subtle abnormalities within the musculoskeletal system. Also, MRI may play a larger role in trauma medicine, particularly with the refinement of open-bore and short-bore technologies. Computed tomography (CT) is an important tool in skeletal imaging because with newer technology the examination can be performed quickly and noninvasively, even in cases of trauma. CT has the ability to define the presence and extent of fractures or dislocations, to assess abnormalities in joints and associated soft tissues, and to help diagnose spinal disorders (Fig. 2-7). Cortical bone gives no signal in MRI, but CT provides ready visualization of bone details and is often used as a follow-up to conventional radiographic imaging for improved detail. Bone tumors, in particular, are now usually imaged with spiral or helical CT because of its excellent ability to display bone margins and trabecular patterns and to assess both bone and soft tissue involvement of tumors. Although CT results in greater contrast resolution compared with radiography, much of the role for imaging other related soft tissues has been usurped by MRI. Quantitative computed tomography (QCT) is also used in evaluating bone mass loss, especially within the vertebral bodies of the spine. Nuclear medicine retains an advantage not offered by either MRI or CT in skeletal imaging: the ability to look at the entire body at one time in a convenient fashion (Fig. 2-8). It allows ready decision making as to whether any pathology shown is an old injury or a new problem, with activity indicating that the bone involved is affected by some new process. In addition, the bone scan is still the standard of care for examination of metastatic processes because it demonstrates metabolic reaction of the bone to the disease process and is more sensitive than comparative radiographic studies. This is also true in many other traumatic or inflammatory diseases of the skeletal system. However, the utilization of positron emission tomography (PET) scanning in skeletal pathologies has started to increase, particularly the use of 18F-NaF (2-deoxy-2-[18F] fluoro-d-glucose). Although primarily used in diagnosing and staging of metastatic disease, it may be appropriate in certain individuals with back pain, to confirm child abuse (especially rib fractures) following abnormal radiographs, and for the diagnosis of osteomyelitis, trauma, inflammatory and degenerative arthritis, avascular necrosis, osteonecrosis of the mandible, condylar hyperplasia, metabolic bone disease, Paget disease, bone graft viability, complications of the prosthetic joints, and reflex sympathetic dystrophy. Osteogenesis imperfecta (OI), sometimes referred to as brittle bone disease, is a quite serious and rather rare heritable or congenital disease affecting the skeletal system. OI is most commonly an autosomal dominant defect. The eight recognized types are classified as type I to type VIII, with type I being the mildest and type VIII the most severe. Prenatal testing of cultured skin fibroblasts (CSF) helps diagnose types II, III, and IV. It is caused by mutations in the two structural genes that encode the α1– and α2-peptides of type I collagen, the main collagen of bone, tendon, and skin. The specific mutations occur in the COL1A1, COL1A2, CRTAP, and LEPRE1 genes. Thus, deficient and imperfect formation of osseous tissue, skin, sclera, inner ear, and teeth is noted in individuals with this disease. The two main clinical groups of OI are based on the age of onset and the severity of the disease. (1) Osteogenesis imperfecta congenita is present at birth. Infants with this disease usually have multiple fractures at birth that heal only to give way to new fractures. This results in limb deformities and dwarfism and may lead to death. (2) In osteogenesis imperfecta tarda, fractures might not appear for some years after birth and then generally stop once adulthood is reached (Fig. 2-9). In some cases, however, a hearing disorder persists because of otosclerosis, which is the formation of abnormal connective tissue around the auditory ossicles. Radiographic evaluation will demonstrate multiple fractures in various stages of healing and a general decrease in bone mass. The bone cortex is thin and porous, and the trabeculae are thin, delicate, and widely separated. The most common inherited disorder affecting the skeletal system is achondroplasia, which results in bone deformity and dwarfism. It occurs in 1 in 15,000 to 40,000 newborns. It is caused by an autosomal dominant gene (FGFR3) at the 4p chromosome location, and this gene does not skip generations. Individuals with this gene have about a 50% chance of transmitting it to their children. Because of a disturbance in endochondral bone formation, the cartilage located in the epiphyses of the long bones does not convert to bone in the normal manner, impairing the longitudinal growth of the bones. Thus, patients with this type of osteochondrodysplasia have a normal trunk size and shortened extremities (Fig. 2-10, A). In some instances, ultrasonography may be used for prenatal diagnosis of achondroplasia. An adult with achondroplasia is usually no more than 4 feet in height, with lower extremities usually less than half the normal length. Additional clinical manifestations of this disorder include extreme lumbar spine lordosis, bowed legs, a bulky forehead with midface hypoplasia (see Fig. 2-10, B), and a narrowing of the foramen magnum within the skull, which causes neural compression. Occasionally, orthopedic surgery may be necessary in the management of complications associated with achondroplasia. The Ilizarov procedure has also been used in an attempt to lengthen the shortened limbs. This procedure was perfected by Dr. Gavriil Ilizarov and has been used for over 30 years. It consists of a corticotomy of the limb, followed by attachment of an Ilizarov fixator, which consists of two circular frames that surround the limb, wires, and rods. By using this method, bones may be made to grow at a rate of approximately 1 mm per day. Bone age radiographic studies may be used to monitor patients with growth hormone (GH) deficiency. These images are analyzed to compare the chronologic age with the radiographic bone age by using one of two methods: (1) the atlas matching method, which was established by Greulich and Pyle in the 1950s; or (2) the point scoring system of Tanner and Whitehouse, which was developed in the 1960s. Most recently, clinical trials have involved GH injections in the treatment of children with achondroplasia. Conclusive results cannot be determined until the children in the current study reach their adult heights. Early research indicates that introduction of GH may have an adverse effect on the cardiovascular system. In addition, these patients may receive genetic and social counseling. Osteopetrosis and marble bone are terms used to characterize a variety of disorders involving an increase in bone density and defective bone contour, often referred to as skeletal modeling. These pathologies involve mutations at the CLCN7 gene. The spectrum of osteopetrosis includes (1) infantile malignant CLCN7-related autosomal recessive osteopetrosis (ARO), which has its onset in infancy; (2) intermediate autosomal osteopetrosis (IAO), which develops in childhood; and (3) autosomal dominant osteopetrosis type II (ADOII), which occurs in late childhood or adolescence. In osteopetrosis, bones are abnormally heavy and compact but nevertheless brittle. The disorders characterizing osteopetrosis include osteoscleroses, craniotubular (affecting the cranium and tubular long bones) dysplasias, and craniotubular hyperostoses. It is important for the technologist to be aware that both the osteosclerotic and craniotubular hyperostotic disorders require an increase in exposure factors to adequately penetrate the bony anatomy because of abnormal bone density (Fig. 2-11). In some cases, adequate radiographic density may never be achieved. All bones are affected, but the most significant changes occur in the long bones of the extremities, vertebrae, pelvis, and base of the skull. Radiographs demonstrate an increase in the density and thickness of the cortex as well as an increase in the number and size of trabeculae, with a marked reduction of the marrow space. A variety of abnormalities of the fingers and toes may occur during fetal development but can be surgically corrected at birth. Failure of the fingers or toes to separate is called syndactyly and causes the physical appearance of webbed digits (Fig. 2-12). It is also associated with Apert syndrome, which is a genetic syndrome involving mutations of fibroblast growth factor receptor 2. Apert syndrome is also responsible for craniosynostosis. Polydactyly (Fig. 2-13) refers to the presence of extra digits, and treatment includes surgical intervention and therapy. A malformation of the acetabulum often results in developmental dysplasia of the hip (DDH). Because the acetabulum does not form completely, the head of the femur is displaced superiorly and posteriorly (Fig. 2-14). Often, the ligaments and tendons responsible for proper placement of the femoral head are also affected. DDH may be unilateral or bilateral and occurs more frequently in females than in males. Other risk factors include a breech position in utero, being the first child, or low levels of amniotic fluid. DDH affects approximately 1 in 1000 births and may be associated with cerebral palsy, myelomeningocele, arthrogryposis, and Larsen syndrome. Larsen syndrome is a mutation of the FLNB gene affecting the production of filament B protein. Sonography may be used to diagnose this anomaly early in life through visualization of the cartilaginous structures of the hip. Conventional radiographs of the hip are often difficult to interpret in the neonate. Radiographic measurements of the anteroposterior (AP) pelvis are obtained and compared with standardized lines. This anomaly should be treated early with immobilization through casting or splinting the affected hip to allow the acetabulum to grow and form a normal joint. If left untreated, this anomaly may result in uneven limb length, hip muscle weakness, and an uneven gait. Scoliosis refers to an abnormal lateral curvature of the spine (Fig. 2-15). Structural scoliosis is associated with vertebral rotation. The lateral curves are usually convex to the right in the thoracic region and to the left in the lumbar region of the spine. Up to 80% of all structural scolioses are idiopathic, although factors such as connective tissue disease and diet have been implicated. Scoliosis does not generally become visually apparent until adolescence. It tends to affect girls more frequently than boys and may cause numerous complications, including cardiopulmonary complications, degenerative spinal arthritis, and fatigue and joint dysfunction syndromes. Nonstructural scoliosis, in which the primary issue is not vertebral rotation, usually results from unequal leg lengths or compensatory postural changes affected by chronic pain elsewhere in the body. A transitional vertebra is one that takes on the characteristics of both vertebrae on each side of a major division of the spine. Most frequently, such vertebrae occur at the junction between the thoracic and lumbar spine or at the junction between the lumbar spine and the sacrum. The first lumbar vertebra and the seventh cervical vertebra may have rudimentary ribs articulating with the transverse processes (Fig. 2-16). A cervical rib most commonly occurs at C7 and may exert pressure on the brachial nerve plexus or the subclavian artery, requiring surgical removal of the rib. Spina bifida is an incomplete closure of the vertebral canal that is particularly common in the lumbosacral area (Fig. 2-17). Often, such patients have no visible abnormality or neurologic deficit, but failure of fusion of the two laminae is visible radiographically (spina bifida occulta). In more severe cases, the spinal cord or nerve root may be involved, which results in varying degrees of paralysis. Treatment of spina bifida is determined on the basis of the extent of the anomaly and requires the services of a variety of physicians. The premature or early closure of any of the cranial sutures is called craniosynostosis. This congenital anomaly causes an overgrowth of the unfused sutures to accommodate brain growth, which alters the shape of the head (Fig. 2-18). It is often associated with Apert syndrome, a genetic disorder that is caused by a mutation of the FGFR2 gene on chromosome 10. Although this defect may be corrected with surgery, brain damage may occur.
Skeletal System
Anatomy and Physiology
Imaging Considerations
Radiography
Magnetic Resonance Imaging
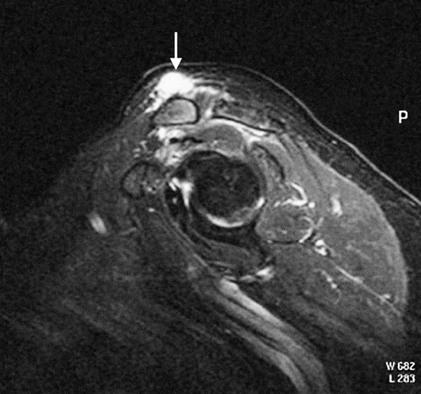
Computed Tomography
Nuclear Medicine Procedures
Congenital and Hereditary Diseases
Osteogenesis Imperfecta
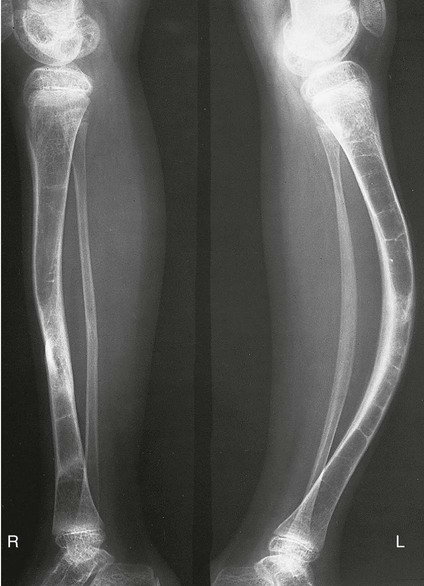
Achondroplasia
Osteopetrosis
Hand and Foot Malformations
Developmental Dysplasia of the Hip
Vertebral Anomalies
Cranial Anomalies
Skeletal System