Fig. 40.1
Pathology slide of a chordoma
Immunohistochemistry shows that almost all tumors stain positively for the epithelial membrane antigen, cytokeratins, S-100 proteins, and are negative for HMB-45 and desmin [4]. One third of all chordomas arise from the skull base, the remainder from the sacrum and the spine. Although they do grow slowly, and rarely metastasize, they are quite often lethal due to their local progression. They commonly involve vital neural or bony structures (Fig. 40.2), thus compromising the effectiveness of surgical or radiation therapy [5]. Intracranial chordomas typically arise from the clivus and can invade the dura, extend in any direction, for example toward the foramen magnum, the petrous bone, the cranio-spinal junction or compress the brain stem, or infiltrate anteriorly the cavernous sinus [6].
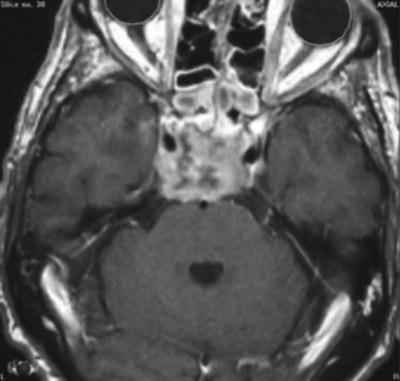
Fig. 40.2
MRI of a skull base chordoma
Chondrosarcomas: General Features
Chondrosarcomas are also rare tumors that can arise from the skull base. Like chordomas, they represent 0.15 % of all intracranial tumors [7] although many series suggest that they are even less common than chordomas [8–10]. Most chondrosarcomas arise de novo although they can occur in Ollier’s disease, Paget’s disease, or in osteochondromas. It is thought that they originate from primitive mesenchymal cells or embryonal remnants of the cartilaginous matrix of the cranium [11] (Fig. 40.3). They are sometimes mistakenly diagnosed as chordoma, but it is quite important to distinguish them from the latter as they have a better prognosis [6, 8, 12–16]. Several subtypes have been proposed that include the hyaline, the myxoid, and mixed hyaline-myxoid; the mesenchymal and dedifferentiated subtypes are less common and disclose a more aggressive behavior, whereas the clear cell subtype is extremely rare [17, 18].
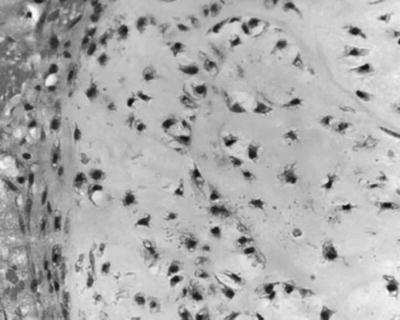
Fig. 40.3
Pathology slide of a skull base chondrosarcoma
On immunohistochemistry, almost all chondrosarcomas stain for S-100 protein, but none for keratin and less than 10 % for epithelial membrane antigens [18]. Thus, tumor markers are helpful adjuncts to differentiate between chordoma and chondrosarcoma: chordomas express cytokeratin and epithelial membrane antigens, whereas chondrosarcomas lack the former and rarely stain for the latter. Like chordomas, chondrosarcomas tend to invade local structures. Distant metastases are uncommon; Hassounah et al. in their literature review found five cases of metastases out of 50 reported patients [19].
Standard Treatment of Chordomas and Chondrosarcomas
Although they have a good number of features in common and their overall management is relatively similar, chordoma and chondrosarcoma of the skull base present with important differences in histopathology, biological behavior, and outcome [6, 18, 20, 21]. For example, in an important series of 109 patients, recurrence and death rates were much higher in patients with chordomas than in patients with chondrosarcomas [6]. Two excellent papers were recently published on chordomas [20] and chondrosarcomas [21], which review the management and outcome of these tumors. There is no question that surgery remains the cornerstone of treatment. The surgeon’s goal should be, whenever possible, to carry out an en bloc resection with a gross total removal [6, 22]. Major advances in surgical techniques and microsurgery in particular have allowed neurosurgeons to perform more macroscopically complete resections, with clearly improved outcomes [22]. However en bloc resections are rarely possible with clival chordoma and thus a maximally safe aggressive surgery with neurological preservation should be the goal [20]. With extensive surgery, Crockard et al. have reported 5-year survival rates of 77 % and 93 %, respectively, for chordoma and chondrosarcoma [15, 16]. Gay et al. obtained a 67 % total or near total resection rate, and for these an 84 % recurrence-free rate was achieved [8].
In spite of these improvements in surgery, it appears that a genuine curative resection with clear margins is not often realistic, and ultimately a majority of patients will recur and die from their disease if no adjuvant radiation is administered. For example, in a series of 13 operated patients with chondrosarcomas from the Netherlands, only three received postoperative photon (2) or proton (1) radiotherapy, and the recurrence-free survival was only 43 % at 5 years [11]. In a systematic literature review of intracranial chondrosarcoma, Bloch et al. found that the 5-year mortality rate was 25 % without and 9 % with postoperative radiotherapy [21].
Therefore, adjuvant, high-dose postoperative radiotherapy has been considered and used for many years both for chordomas and chondrosarcomas. In spite of old claims that chondrosarcomas and chordomas are radioresistant tumors [23], conventional external beam radiotherapy was shown to provide useful and prolonged palliation in overt residual disease [5]. It was however suggested for a long time that these tumors, in order to be controlled, need to receive relatively high-dose radiotherapy and that the total dose should be increased to 70–80 Gy [24–27]. Currently, with high-precision external beam photon radiotherapy, the dose to the PTV can be escalated to 65–70 Gy, with local control rates comparable to those obtained with particle therapy [26].
Because of the presence of neighboring critical structures like the brain stem or the temporal lobe, with older techniques the total dose was limited to 50–60 Gy, which was clearly insufficient to control even microscopic residual disease in the long term. Charged particles such as protons, helium, or neons or carbon ions are well suited for extremely precise localization of radiation and permit an increase of the total dose from 15 to 35 % compared with conventional X-rays [10]. In their experience with charged particle irradiation of tumors of the skull base, Castro et al. have treated 53 chordomas and 27 chondrosarcomas, from 1977 to 1992, with a mean dose of 65 Gray equivalent (GyE) (range, 60–85 GyE) [10]. Five-year local control was 63 % for chordomas and 78 % for chondrosarcomas [10]. Since then, a number of patients were treated with particle therapy, generally proton beam therapy in the United States, France, Japan, Switzerland, and Germany, with protocols using high doses of irradiation.
At the Massachusetts General Hospital (MGH), where chordomas and chondrosarcomas were treated with 160 MV proton beams, with a median dose of 69 CGE (Cobalt-Gray-Equivalent), it was found that chordomas had a 31 % rate of failure of which 95 % were local [24, 25]. It was also shown that the 10-year local control was the highest with chondrosarcomas, followed by male chordomas and female chordomas, with 94 %, 65 %, and 42 % local control rates, respectively [14]. At Loma Linda University, 58 patients with chordomas and chondrosarcomas received proton beam radiotherapy with a mean dose of 70 CGE, with a local control rate of 92 % for chondrosarcomas and 78 % for chordomas [13]. An update of the French series at Orsay on 100 patients confirmed their previous results with a combination of photons and proton beams (201 MV with a median dose of 67 CGE). In this report, the 2- and 4-year local control rates were 86 % and 54 %, respectively [9]. Patients with chordomas treated with proton beam therapy at Tsukuba University received a median dose of 72 Gy with 5-year local control and local-specific survivals of 42 % and 72 %, respectively [27]. At the Paul Scherrer Institute, Switzerland, 64 patients with skull base chordomas (42) and chondrosarcomas (22) were treated with the spot-scanning-based proton radiotherapy technique with a median dose of 75.5 Gy for chordomas and 68.4 Gy for chondrosarcomas [28]. Actuarial 5-year control rates were 81 % for chordomas and 94 % for chondrosarcomas; the 5-year freedom from high-grade toxicity was 94 % [28]. With carbon ion radiotherapy, the experience from Heidelberg and GSI in Darmstadt on 96 chordoma patients disclosed a 70 % local control and 88 % survival at 5 years [29]. The same group obtained a 90 % 4-year local control and 98 % 5-year survival in a series of 54 patients with chondrosarcomas [30]. In the experience with proton beam and carbon ion radiotherapy, late grade 3–4 toxicities were generally low, given the high radiotherapy dose delivered, and were reported to be between 4.5 and 7 % [13, 25, 28–31]. In conclusion, high-precision, high-dose particle radiotherapy is followed by an excellent local control for chondrosarcoma and a reasonably good local control for chordoma, with a low toxicity. Postoperative proton beam radiotherapy is often considered now as the standard treatment for these tumors. We will examine next if high-precision and high-dose STRT can be considered as alternative treatments to proton beam therapy.
Fractionated Stereotactic Radiotherapy of Chordomas and Chondrosarcomas
Patient Selection
As previously emphasized, the standard management of chordomas and chondrosarcomas should include a maximal surgical resection, followed by postoperative high-precision, high-dose radiotherapy, if one wants to maximize the chances of local control [6, 15, 16, 20–30].
The indications for STRT are essentially the same as those for RS (see chapter on RS), except that STRT combines the precision of stereotactic positioning with the radiobiological advantage of fractionation, especially in large tumors [12]. Thus, in tumors larger than 30 mm in diameter, there is an advantage of using STRT rather than RS, at least in theory. In the series from Heidelberg, where STRT was used for chordomas and chondrosarcomas of the skull base, the median target volume was 56 cm3 (range, 17–215 cm3) for chordomas and 102 cm3 (range, 24–237 cm3) for chondrosarcomas [12]. This is in sharp contrast with the mean or median volumes described in the RS series, which ranged from 4.6 cm3 (range, 0.98–10.8 cm3) to 14.6 cm3 (range, 2.9–52 cm3) [32–36].
Treatment Techniques
Planning techniques are similar to those used for any RS or STRT of other intracranial lesions. Immobilization systems including various stereotactic head frames (RS) or relocatable head masks (STRT) are used for imaging studies under stereotactic guidance and for treatment. All patients should undergo high-resolution computed tomography (CT) and if not contra-indicated magnetic resonance imaging (MRI), with image fusion for treatment planning. In both cases, ultrathin slices are required. With regard to fractionated STRT, the Heidelberg group has defined the clinical target volume (CTV) as the visible tumor on CT and MRI and the potential residual tumor, taking preoperative imaging into account [12]. They added a 2-mm safety margin for the planning target volume (PTV). The median prescribed dose at isocenter was 66.6 Gy for chordomas and 64.3 Gy for chondrosarcomas, with a median daily dose of 1.8 Gy [12]. More recently, Boguci et al. have used image-guided intensity-modulated fractionated STRT, with a median dose of 66.6 Gy prescribed at the 90 % isodose, so that the dose at the isocenter was 74 Gy [37]. Figure 40.4a, b shows an example of linac-based STRT treatment plan at our institution (CHUV) for a 59-year-old patient who was not operated due to important comorbidities. The patient, with a skull base chordoma, received a total dose of 63 at 1.8 Gy per fraction, using a micro-multileaf collimator with six fixed beams from a 6-MV linear accelerator.
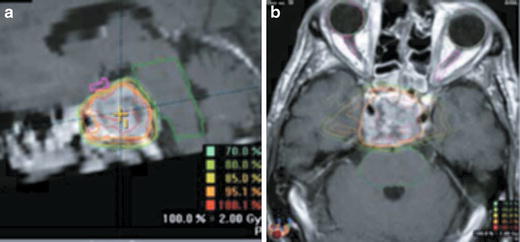
Fig. 40.4
Treatment plan for STRT of a case of chordoma (a) sagittal (b) axial
Treatment Outcomes
Because of the rare occurrence of chordomas and chondrosarcomas, the published experience with either RS or STRT is still limited. Table 40.1 summarizes some recent series with RS and STRT for chordomas and Table 40.2 the experience with chondrosarcomas. As can be seen, these series of Gamma Knife radiosurgery (GKRS) and STRT have gathered a limited number of cases of chordomas [12, 32, 33, 36–40] and very few cases of chondrosarcomas [12, 32–34, 36].
Table 40.1
Treatment outcomes, chordomas
Series | Year | No. of patients | Technique | Median prescribed dose (Gy) | 5-year local control (%) | 5-year survival (%) | Median follow-up | Comments |
|---|---|---|---|---|---|---|---|---|
Krishnan et al. [33] | 2005 | 25 | GKRS | 15 | 55 | 4.8 years | EBRT also used in a proportion of patients | |
Martin et al. [32] | 2007 | 18 | GKRS | 16 | 63 | 63 | 7.7 years | EBRT also used in a proportion of patients |
Hasegawa et al. [36] | 2007 | 27 | GKRS | 14 | 72 | 84 | 6 years | |
Dassoulas et al. [38] | 2009 | 15 | GKRS | 12.7 | 42 | 80 | 5.8 years | LC improved to 50 % after 2nd GKRS |
Kano et al. [39] | 2011 | 71 | GKRS | 15 | 66 | 74 | 5 years | NAGKC (6 centers) |
Henderson et al.[40] | 2009 | 18 | CyberKnife | 35 | 59 | 82 | 4 years | 35 Gy in 5 sessions |
Debus et al. [12] | 2000 | 37 | Linac STRT | 66.6 | 50 | 7682 | 19 months | 1.8 Gy per fraction |
Bugoci et al. [37] | 2012 | 12 | Linac STRT | 66 | 38 | 76 | 3.5 years | 2 Gy per fraction |
Table 40.2
Treatment outcomes, chondrosarcomas
Series | Year | No. of patients | Technique | Median prescribed dose (Gy) | 5-year local control | 5-year survival | Median follow-up | Comments |
|---|---|---|---|---|---|---|---|---|
Krishnan et al. [33] | 2005 | 4 | GKRS | 15 | 100 % | 4.8 years | ||
Feigl et al. [34] | 2005 | 10 | GKRS | 17 | 100 % | NA | 17 months | One patient had 2 GKRS |
Martin et al. [32] | 2007 | 10 | GKRS | 15 | 88 % (6 years) | 88 % | 7.7 years | 1.8 Gy per fraction |
Hasegawa et al. [36] | 2007 | 7 | GKRS | 14 | 80 % | 83 % | 6 years | |
Debus et al. [12] | 2000 | 8 | Linac STRT | 64.9 | 100 % | 100 % | 19 months |
Overall, these results confirm the known differences in outcome between chordomas and chondrosarcomas. Results on local controls and survivals are fairly similar to those obtained after postoperative proton beam radiotherapy. For chordomas, the 5-year local control rates varied between 38 and 72 %, whereas for chondrosarcomas, it was between 80 and 100 %. The rather wide variations between the different series are probably due to differences in surgical techniques, tumor size, and in the patients’ populations, some reports having included both primary treatments and treatments for recurrence. The experience at the University of Heidelberg and at the Kaiser Permanente in Los Angeles with STRT showed encouraging local control rates, which were comparable to those obtained with RS [12, 37]. It should also be re-emphasized that patients treated by STRT had on average a much larger residual tumor volume compared with those who received GKRS (vide supra). STRT yields the same excellent local control rates for chondrosarcomas as with RS, but admittedly the number of patients was quite low [12].
Functional outcome after RS was reported in the rather large North America Gamma Knife consortium: of 57 patients with prior neurological deficits, 17 improved, 18 were unchanged, and 16 deteriorated [39].
Radiation-induced complications are generally moderate; however, cranial nerve dysfunction, pituitary insufficiency, and occasionally brain necroses were reported [32, 33]. Similarly, the complication rates after STRT are quite low: from the 45 patients treated with STRT in Heidelberg, only one presented with symptomatic infarction of the pons [12].
Conclusion
In conclusion, RS, either with GKRS or linac RS, and STRT appear to be safe treatments as postoperative complement to surgery or for recurrent chordomas and chondrosarcomas of the base of the skull. It is thus reasonable to state that, in the absence of a nearby proton beam therapy facility, fractionated STRT represents a possible, safe and cheaper alternative treatments for these skull base tumors.
Paragangliomas
General Features
PGLs are neuroendocrine tumors originating from the autonomic nervous system derived from neural crest cells. Approximately 80–85 % of these tumors are located in the adrenal medulla and called pheochromocytomas (PCCs), whereas 15–20 % grow in the extra-adrenal chromaffin tissue and are referred to as secreting paraganglioma (sPGLs). These tumors are rare and occur in 2–8 per 1,000,000, with a peak incidence in the third to the fourth decade of life, with an almost equal distribution between female and male patients; familial tumors occur at an earlier age [41]. They are generally slow growing, highly vascular, with a high intracellular liquid content and with frequent intratumoral cysts [42–45] (Fig. 40.5). Depending on their origin, PGLs can be divided into two groups: sympathetic and parasympathetic tumors. PCCs fall into the former category. Sympathetic PGLs can be found anywhere along the sympathetic ganglia, in the thorax, abdomen, and pelvis, and they tend to hypersecrete catecholamines, like adrenal PCC. In contrast, parasympathetic PGLs are principally located in the head and neck region (HNPGLs) and base of the skull, and in their majority are nonsecretory. They can be multicentric, especially in familial cases [45]. In some instances, they can reach very large sizes and are referred to as giant tumors [45]. One quarter of PGLs are malignant, defined by the presence of chromaffin tissue in sites where it is normally absent, such as lymph nodes, bone, liver, and lung [41]. Unfortunately, there are currently no consistent methods to differentiate benign from malignant forms, and malignancy is revealed by distant metastases [46]. The clinical behavior of PGLs is generally indolent with long time intervals between the first symptom and the diagnosis [42, 45]. In case of sPGLs, hypertension is the main symptom, which can be continuous, intermittent, or paroxysmal. HNPGLs are characterized by a mass effect or the infiltration of adjacent anatomical structures such as the temporal bone, the middle ear, the clivus, the jugular vein, the internal carotid artery, the cavernous sinus, the hypoglossal canal, and cranial nerves V–XII [43, 44, 47–52] (Fig. 40.6). A palpable neck mass, as well as dysphagia, pain, tinnitus, or cranial nerve palsies can also occur [52]. The several classifications take into account tumor location, extent, and size. The Fisch classification is one of the most commonly used [50]. The clinical behavior of malignant PGLs may also be associated with systemic symptoms, including fatigue, weight loss, and anorexia, or clinical manifestations due to metastatic disease, such as bone metastases. Metastases can occur at the outset or after many years. Population-based studies have established that approximately one third of patients having sporadic PGL have a germline mutation in a known susceptibility gene [53–56]. Ten known susceptibility genes for PGL have been identified to date: three genes for three distinct cancer susceptibility syndromes, VHL for von Hippel–Lindau disease, NF1 for Neurofibromatosis Type 1, and RET for Multiple Endocrine Neoplasia Type 2 (MEN2); others comprise the succinate dehydrogenase (SDH) complex subunit genes (SDHA, SDHB, SDHC, SDHD), the SDH complex cofactors (SDHAF2), and two newly recognized susceptibility genes, TMEM127 and MAX [57]. SDHB mutations are usually linked to a higher morbidity and mortality than mutations in the other SDH genes [58]. A recent meta-analysis of studies concerning SDHB-mutated patients has emphasized that 31 % of their tumors were malignant [59]. Notably, VHL- and SDH-mutated PGLs are associated with angiogenesis, hypoxia, and reduced oxidative response [60] while the group containing RET- and NF1-mutated tumors is linked to an abnormal activation of kinase-signaling pathways, such as RAS/RAF/MAPK and PI3K/AKT/mTOR [61, 62]. Also TMEM127- and MAX-mutated tumors have been associated with the activation of mTOR-signaling pathway [63, 64]. The recommended screening test for initial evaluation of PGLs is the measurement of plasma-free metanephrines or urine-deconjugated differential metanephrines [65]. Metanephrines have a higher sensitivity, ranging around 98–99 %, in comparison to plasma or urine catecholamines and vanilmandelic acid [66, 67]. The biochemical phenotype does not allow to differentiate malignant from benign PGLs. However, the presence of predominantly noradrenaline-producing PGLs and high levels of plasma dopamine or its metabolite methoxytyramine may suggest malignancy [66, 68]. Plasma chromogranin A (CgA), which is a protein stored and cosecreted with catecholamines, is frequently increased in functioning and non-functioning PGLs [69]. CgA shows a sensitivity of 83–89 %, and very high plasma levels of CgA are generally related to malignancy [66]. Computed tomography (CT) and magnetic resonance (MRI) are major tools for the first imaging approach in patients with PGLs. MRI shows a good accuracy, with a sensitivity of 90–100 % and specificity of 50–100 %, especially for the detection of extra-adrenal disease [70]. Ultrasound is used for the detection of HNPGLs, whereas for other disease sites its role is limited [70, 71]. Concerning functional imaging, 131I- or 123I- metaiodobenzylguanidine (MIBG) scintigraphy has been used as the first-line nuclear medicine technique but 123I-MIBG is superior to 131I-MIBG, with a sensitivity of 83–100 % and a specificity of 95–100 % [70]. Regarding PET imaging, somatostatin analogues labelled with gallium-68 can be used. This tracer seems to be superior to 18F-labelled fluorodeoxyglucose (18F-FDG) in identifying malignant sPGLs [72].
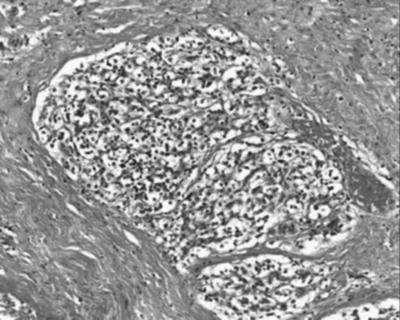
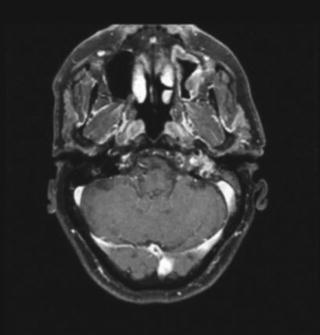
Fig. 40.5
Pathology slide of a case of paraganglioma
Fig. 40.6
MRI of a case of paraganglioma
Standard Treatment for Paragangliomas
The treatment of choice of PGL is a matter of controversy. Surgery has an important role but it is by far not the only option. With recent imaging tools, innovative surgical techniques, and cranial nerve monitoring, results have improved regarding the possibility of complete removal, and immediate complications have decreased. However, like for chordomas and chondrosarcomas of the skull base, a genuine total resection without sacrifice of cranial nerves is rarely possible. Therefore, postoperative cranial nerve damage can be quite substantial, with potential impairment of nerves VII and IX–XII [45, 48, 49]. For example, in one series, preoperative deficit of cranial nerves X–XII was between 20 and 30 % and postoperative deficits increased to 25–50 %, although facial function recovery was observed in 95 % of patients [49]. The rate of complete surgical removal is quite variable and is claimed to be between 40 and 96 % [48, 49]. In general, data regarding resection rates and the actual occurrence of complications are difficult to interpret due to the heterogeneity and limited size of most series. There are only few data on local control and long-term survival after surgery. Some surgeons have claimed that in their hands, the recurrence rate was quite low. Nora et al. have reported on 59 carotid body tumors, of which only 3 (6 %) presented with a recurrence after surgery, and only one developed metastatic disease [73]. Pareschi et al. in their series of 37 glomus jugulare tumors, of which 96 % had a complete resection, found no relapse, but the mean follow-up was only 4.9 years [48]. Amongst 28 patients with the so-called complex glomus jugulare tumors, Al-Mefty and Teixeira were able to perform a complete resection in 24 patients and observed altogether two recurrences [45]. However, in other studies, local control was much lower, with 70–100 % recurrence rates after surgery alone [74, 75]. Because these tumors tend to recur after many years, the actual failure rates are underestimated, especially with short follow-ups. In addition many patients are lost to follow-up [45, 48]. Long-term mortality due to recurrence after surgery is not so trivial and was reported to be between 5 and 13 % [76, 77]. Like for chordoma and chondrosarcoma, some authors have claimed that PGLs are radioresistant and that radiotherapy was associated with long-term complications [45]. These assertions were based on old studies in which ancient orthovoltage techniques were used. Indeed, with very conformal linac-based techniques, fractionated external beam radiotherapy represents an excellent alternative, or sometimes a complementary treatment to surgery. Cole et al. have treated 32 tumors of the glomus jugulare or glomus vagale with megavoltage units with doses of 45 Gy in 5 weeks [78]. Their very long-term results have yielded a local control rate of 94 % at 10 years [78]. Konefal et al. have treated 26 patients with 45–50 Gy; amongst these, 15 of 16 glomus tympanicum and four of six glomus jugulare tumors achieved a long-term control, with a mean follow-up period of 10.5 years [79]. In a large series, 80 PGLs of the temporal bone, carotid body, or glomus vagale were treated by radiotherapy alone (72) or postoperatively (8) [80]. Local control was 94 % and only 5 local recurrences were observed between 2.6 and 18.8 years, of which two could be salvaged by surgery [80]. Other groups could obtain good local controls with exclusive or postoperative radiotherapy [74, 75]. Powell et al. have treated 46 patients with glomus jugulare or glomus tympanicum tumors with doses between 45 and 50 Gy and a 75 % actuarial control was achieved at 25 years [75]. Notably, in these radiotherapy series, between 18 and 30 % were previously, and sometimes heavily pretreated by surgery. Contrarily to older reports, complication rates were low with the most recent radiotherapy techniques [78–80]. Previously, it was thought that these tumors were “radioresistant” because after irradiation, they shrink but rarely disappear completely on follow-up [78]. Therefore, local control after radiotherapy should be defined as the absence of clinical and radiological progression. Springate et al. have reviewed the outcome of PGL after various treatments [81]. They noted that local control after surgery, surgery combined with radiotherapy, or radiotherapy alone was 86 %, 90 %, and 92 %, respectively. In the same review, treatment-related morbidity after surgical excision was frequent, whereas late complications were rare after radiotherapy [81]. This was more recently confirmed by Huy et al. who compared in a retrospective and comparative analysis radiotherapy and surgery in 88 cases of jugular PGLs, in terms of function and tumor control [82]. Forty-seven patients with type C or D jugular PGLs underwent surgery after endovascular embolization between 1984 and 1998, and were followed up on average during 66 months. Forty-one patients with type C jugular PGLs were treated by external beam conformal radiotherapy between 1998 and 2003 with a total mean dose of 45 Gy (range, 44–50 Gy); the mean follow-up was 50 months [82]. This study demonstrated that with radiotherapy, a 96 % local control was achieved, whereas with surgery it was 86 % [82]. After surgery, postoperative complications included dysphagia, aspiration, and facial paralysis. Patients treated by radiotherapy developed only minor sequelae. In a British retrospective study, 21 patients with PGL of the head and neck region received fractionated external beam radiotherapy with a median dose of 50 Gy in 30 fractions [83]. With a median follow-up of 55 months, a 92 % 5-year local control rate was observed [83]. Another retrospective study, with a median follow-up of 9 years, demonstrated that treatment of PGL by external beam radiotherapy yielded a 5-year local control rate of 96 % and a 10- and 15-year local control rate of 90 % [84].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


