Spine
Teresa Chapman, MD
LEARNING OBJECTIVES
1. Describe the significance of an abnormal cutaneous finding in the midline lower back of a neonate, and describe the appropriate imaging workup.
2. Discriminate between lipomyelocele and lipomyelomeningocele on MRI.
3. Name the brain malformation associated with myelomeningocele.
4. Recognize spondylolysis on spine radiography.
5. Define Scheuermann kyphosis.
6. Provide a differential diagnosis for an aggressive lytic lesion involving the posterior elements of the spine.
7. List two possibilities for a lytic lesion in the anterior spinal column.
8. Localize spinal tumor by anatomic compartment, and list two diagnostic considerations.
Introduction
Pediatric spine disorders include a wide number of pathophysiologic processes, including congenital malformations, heritable spinal muscular atrophy, malignancy of the vertebral column and of the cord, inflammatory diseases, and trauma. This chapter focuses on the appropriate imaging evaluation and clinical implications of spine disorders that are predominantly seen in the pediatric population. Scoliosis, which is a common pediatric spine abnormality and affects approximately 2% to 3% of the population in the United States,1 is discussed in Chapter 28.
An essential discussion of imaging studies of the pediatric spine begins with the embryology of this complex structure. The first phase of spine development, gastrulation, begins in the second and third weeks of gestation. During this time, the two layers of the embryonic plate divide into three definitive layers—the ectoderm, mesoderm, and endoderm. The notochordal process forms between ectoderm and endoderm, with inward migration of prospective notochordal cells.2 During the second phase, called primary neurulation, occurring in the 3rd and 4th weeks of development, the spinal cord develops through an interplay of cell-signaling factors steering the ventral and dorsal neural elements. A widely taught theory argues that the primitive neural plate closure begins in a bidirectional fashion in the midthoracic region and extends cephalad and caudad. Toward the end of neurulation, the neural tube is still open at the cranial neuropore and at the caudal neuropore, and these normally close between the 25th and 27th days of embryologic development. Disruption of primary neurulation, then, would lead to neural tube defects in either the craniocervical or lumbosacral region.2,3 The subsequent process of secondary neurulation forms the lower half of the sacrum, the coccyx, and the corresponding sacrococcygeal spinal cord metameres (the origins of the conus medullaris and the filum terminale).2,3
Subsequent to primary and secondary neurulation, there is ascent of the spinal cord, attributable to the relatively faster growth of the vertebral column than the cord.2 The spinal cord terminates at or above the inferior aspect of the L2 vertebral body in 95% of the adult population, and it terminates at or above the L1-L2 disc space in 57% of the population.3 The conus medullaris has reached its mature adult level in most term infants and in 100% of cases by age 3 months.4,5 Therefore, a conus terminating above the level of the L2-L3 disc space is considered normal (Fig. 36.1). A low conus termination is concerning for tethered cord, which is a clinical syndrome involving progressive neurologic deterioration, such as motor and sensory dysfunction of the lower extremities, muscle atrophy, urinary incontinence, and orthopedic deformities such as scoliosis or foot and hip deformities.6 Early surgical intervention is the standard of care for these patients to avoid progression of neurologic deterioration and late irreparable damage.7,8
NEURAL TUBE DEFECTS
Spinal dysraphism (from the Greek terms, dys = bad, rhaphe = suture) is a broad term that refers to incomplete closure of the posterior elements. It is estimated that spinal dysraphism has an incidence of approximately 0.05 to 0.25 per 1,000 births.9,10 Such anomalies may be divided into skin-covered (closed spinal dysraphism [CSD]) and uncovered (open spinal dysraphism [OSD]) pathologies. Imaging modalities to evaluate these entities
commonly include spine radiography, sonography, and MR imaging. MR is appropriate when a subcutaneous mass is present and when surgical therapy is clearly required, such as with OSD or CSF drainage from a skin dimple or sinus tract. Occasionally, CT is appropriate for definition of bony anatomy, usually in the context of surgical planning.
commonly include spine radiography, sonography, and MR imaging. MR is appropriate when a subcutaneous mass is present and when surgical therapy is clearly required, such as with OSD or CSF drainage from a skin dimple or sinus tract. Occasionally, CT is appropriate for definition of bony anatomy, usually in the context of surgical planning.
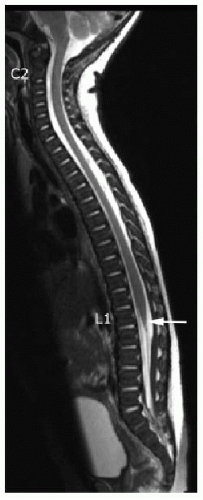 FIG. 36.1 • Normal conus terminus. Sagittal T2-weighted MR image of total spine in an 8-weekold infant shows normal cord and normal cord terminus (arrow) at the level of L1-L2. |
Closed spinal dysraphism
The term “occult spinal dysraphism” is a historically used synonym for CSD, although some experts discourage the use of the term “occult” since most CSD cases have cutaneous stigmata in the midline lower back that readily suggest an underlying spine anomaly.11 These cutaneous signs include hairy nevus, capillary hemangioma, complex sacral dimples, cutaneous dystrophy, patches of discoloration, caudal appendage, and subcutaneous masses. When cutaneous anomalies are present, an underlying spine anomaly is present in approximately 50% of cases.12,13 In contrast, a cutaneous finding that is quite common in the normal population is the simple sacral dimple, defined as a solitary, nondraining dimple less than 5 mm in diameter and low in the gluteal cleft (less than 2.5 cm from the anus).13 The risk of a clinically significant underlying spine anomaly in the setting of a simple sacral dimple is exceedingly low.14
Imaging evaluation for a suspected CSD, based on the presence of a complex dimple—one that is greater than 5 mm in diameter, located 2.5 cm above the anus, deep, or draining fluid—or any other concerning cutaneous lesion, begins with spine sonography, often combined with radiography. The purpose of imaging is to determine the level of conus termination and to screen for any associated finding, such as filum terminale thickening or an intrathecal lesion such as a congenital intrathecal lipoma.
Spine sonography must be done before the dorsal bone elements of the spine have fused, and therefore, appropriate timing is prior to 4 months of age.15, 16, 17 and 18 Sonography is performed with the infant in prone position. A linear high-frequency transducer is used. The normal distal cord appears smoothly demarcated and tapers to the apex of the conus. Anechoic CSF should be present dorsal to the cord. Centrally within the cord parenchyma is an echogenic line representing the central canal (Fig. 36.2A). The nerve roots of the cauda equina float freely within the subarachnoid CSF space and appear echogenic distal to the cord termination (Fig. 36.2B, C). Lack of cauda equina movement with CSF pulsation implies tethering. The filum terminale is a linear echogenic structure that normally measures less than 2 mm in thickness and extends from the conus to the end of the thecal sac (Fig. 36.2C).17,18
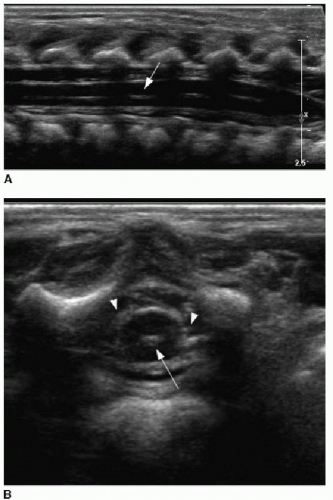 FIG. 36.2 • Normal spine ultrasound. A: Longitudinal view of spine in a 10-day-old infant shows normal appearance of distal cord. Linear central echogenicity (arrow) reflects the central canal. Apparent disruption of canal is due to posterior acoustic shadowing by posterior vertebral osseous elements. B: Axial view of distal cord shows punctate echogenic central canal (arrow) and floating nerve roots of cauda equina adjacent to cord (arrowheads). C: Longitudinal view of lower spinal canal showing echogenic roots of cauda equina (arrowheads). Distal cord tapers into conus (white arrow). Filum terminale (black arrow) extends from conus to distal thecal sac. D: Anteroposterior supine radiograph of spine following sonographic identification of conus with radiopaque marker placed on skin at time of ultrasound. Radiograph shows conventional vertebral body numbering and radiopaque marker at L1 level, confirming normal conus position. |
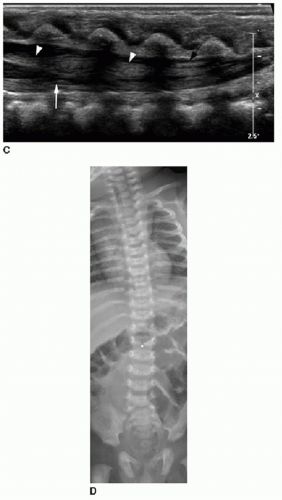 FIG. 36.2 • (Continued) |
Position of the conus can be established by using bony landmarks such as the last rib indicating T12 and the lumbosacral lordosis indicating L5-S1 (Fig. 36.3). Determining the vertebral body levels sonographically can be challenging, and some centers will mark the conus level on the skin at the time of sonography using a radiodense marker and obtain a follow-up spine radiograph (Fig. 36.2D). This allows for definitive confirmation of the conus level and also provides an opportunity to assess the bony elements for anomalies.
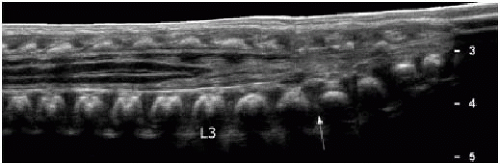 FIG. 36.3 • Abnormally low-lying conus identified by sonograp hy. Longitudinal ultrasound of lower spine in a 2-day-old infant shows lumbosacral lordosis (arrow), assumed to reflect location of the L5-S1 disc space. Conus termination is thereby identified at L3 level, abnormally low (later confirmed by spine MR imaging, not shown here). |
Anatomic variants of the distal spine may be observed sonographically, and these are considered clinically insignificant in the setting of a normal conus termination.18 These include the following: (1) a fatty filum terminale—if the filum is thickened more than 2 mm and echogenic; see Figure 36.4A and B; (2) a ventriculus terminalis, which is due to incomplete regression of the embryonic terminal ventricle in the conus medullaris and is notable for its dorsal convexity, straight ventral aspect, and sharply tapered margins (Fig. 36.4C); and (3) a filar cyst, characterized
by a location immediately distal to the conus in the midline, with anechoic contents and a fusiform shape (Fig. 36.4D).
by a location immediately distal to the conus in the midline, with anechoic contents and a fusiform shape (Fig. 36.4D).
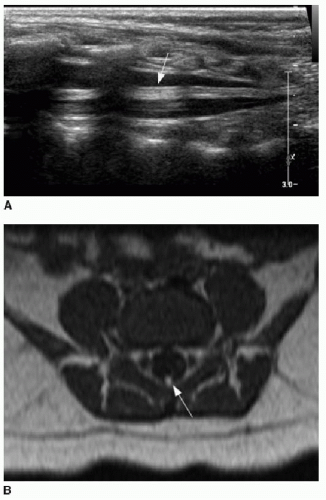 FIG. 36.4 • Spine anatomic variants identified by spine sonography in neonates. A: Longitudinal sonographic view of lower thecal sac in an 8-week-old infant with sacral dimple shows abnormally echogenic and mildly thickened filum terminale (arrow), measuring 2.2 mm thick. B: Axial T1-weighted MR image of same infant shows abnormal T1 hyperintensity of filum (arrow), consistent with fatty filum. Conus termination was normal (not shown here). C: Longitudinal sonographic view of spine in a 12-day-old infant with sacral dimple shows mild anechoic dilatation of the terminal central canal (arrow) with a configuration typical of ventriculus terminalis. D: Longitudinal sonographic view of spine in a 2-day-old infant with sacral dimple shows a small anechoic elongated cyst (arrow) associated with the filum terminale at the distal cord (conus annotated by arrowhead), consistent with a filar cyst. |
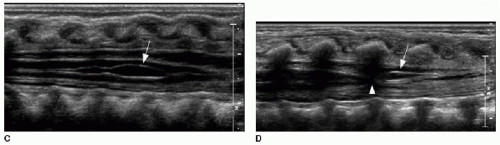 FIG. 36.4 • (Continued) |
The presence of a subcutaneous mass on the patient’s back indicates a more complex CSD. The two most common closed dysraphic entities presenting with a lumbar midline subcutaneous mass are lipomyelocele and lipomyelomeningocele.3 The mass is associated with a subcutaneous lipoma in both of these entities. Differentiating between these two on MR imaging requires identification of the interface between the lipoma and the neural placode. The placode is a segment of nondifferentiated embryonic neural tissue, abnormally halted at the neural plate stage. On MR imaging, the neural placode appears as a small flat, bandlike, isointense segment of tissue. With a lipomyelocele, the lipoma-placode interface will be positioned within the spinal canal, as opposed to the lipomyelomeningocele, which involves outward ballooning of the subarachnoid spaces through the dorsal osseous spinal defects, displacing the lipoma-placode interface outside the spinal canal (Fig. 36.5).2,3,11
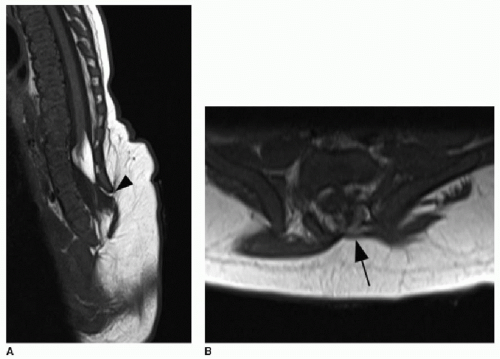 FIG. 36.5 • Lipomyelocele and lipomyelomeningocele on MRI in two different patients. Sagittal (A) and axial (B) T1-weighted images through lower spine in a 3-month-old infant show lower spine dysraphism and absence of dorsal osseous elements (black arrowhead). Conus position is low and terminates in neural placode mixed with an abnormal T1 hyperintense mass, consistent with lipoma, in lower thecal sac. Lipoma-placode interface within spinal canal (black arrow) classifies this as lipomyelocele. C: Sagittal T1-weighted image of a different 3-month-old infant with closed spinal dysraphism shows low termination of conus and neural placode intermixed with fat signal extending dorsal to the dysraphic defect. D: Axial T2-weighted image from the same case shows cystic component (white arrow) outside of spinal canal. Fat signal (white arrowhead) is partially within spinal canal. E: Axial T1-weighted image shows lipoma-placode interface partially outside spinal canal (arrow). These findings indicate lipomyelomeningocele. |
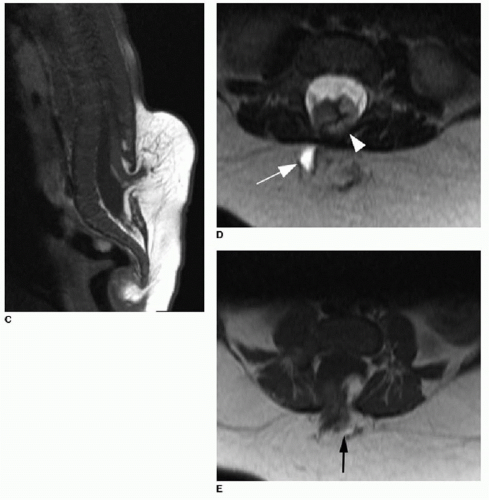 FIG. 36.5 • (Continued) |
Two other forms of CSD in the lumbosacral region that are much less common do not have a fatty component. These are the meningocele, characterized simply by meninges ballooning out through the dorsal spina bifida, and the terminal myelocystocele, which is rare and is characterized by syringomyelia within a low-lying cord that balloons outward through the osseous defect along with meninges (Fig. 36.6).11
Diastematomyelia
Split cord malformations have been referred to by the terms diastematomyelia, meaning cleft cord, and diplomyelia, meaning duplicated cord. This type of malformation represents approximately 4% of all CSD cases.3,11 This anomaly stems from very early failure of the paired parasagittal notochordal origins to fuse in the thoracic region.19 Type I is characterized by the presence of a bony bar separating the two cord segments each within a distinct dural sleeve, whereas in type II, both hemicords are contained within the same thecal sac19,20 (Fig. 36.7). Diastematomyelia is strongly associated with the presence of a hairy patch (hypertrichosis) in the midline back, particularly type I.20, 21 and 22 The affected level is usually in the lower thoracic or lumbar spine, although high thoracic and cervical cases do rarely occur.20, 21 and 22 Associated abnormalities include syrinx, scoliosis, foot deformities, lower extremity weakness, and bowel and bladder sphincter dysfunction.20, 21 and 22
Caudal Regression Syndrome (Caudal Agenesis)
Caudal agenesis encompasses a spectrum of anomalies characterized by partial agenesis of the lower spinal column, anal imperforation, and genitourinary system anomalies.23 The most severe cases include lower limb dysplasia or agenesis, termed sirenomelia. As a group of disorders, caudal agenesis accounts for approximately 16% of all CSD cases.11 Caudal agenesis is associated with maternal diabetes mellitus.24,25 Agenesis of the sacrococcygeal
spine may be part of syndromic associations such as VACTERL/VATER (vertebral anomaly, anorectal malformation, cardiac anomalies, tracheoesophageal fistula, renal anomalies, limb deformities), OEIS (omphalocele, cloacal exstrophy, imperforate anus, and spinal deformities), and the Currarino triad (partial sacral agenesis, anorectal malformation, and presacral mass, such as hamartoma, anterior meningocele, or sacrococcygeal teratoma).26, 27 and 28 On imaging, the lower spine will be deficient, and the lower cord is truncated (Fig. 36.8).
spine may be part of syndromic associations such as VACTERL/VATER (vertebral anomaly, anorectal malformation, cardiac anomalies, tracheoesophageal fistula, renal anomalies, limb deformities), OEIS (omphalocele, cloacal exstrophy, imperforate anus, and spinal deformities), and the Currarino triad (partial sacral agenesis, anorectal malformation, and presacral mass, such as hamartoma, anterior meningocele, or sacrococcygeal teratoma).26, 27 and 28 On imaging, the lower spine will be deficient, and the lower cord is truncated (Fig. 36.8).
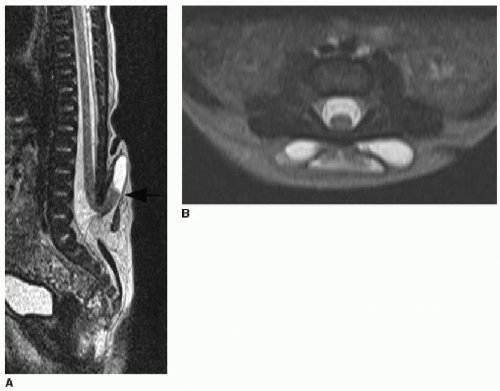 FIG. 36.6 • Terminal myelocystocele. Sagittal (A) and axial (B) T2-weighted MR images of lower spine in a 7-day-old infant presenting with midline lower back skin-covered mass and hairy patch. There is dysraphism of the lower spine through which the terminal cord protrudes. Cord terminus expands into a cyst (black arrow in A). |
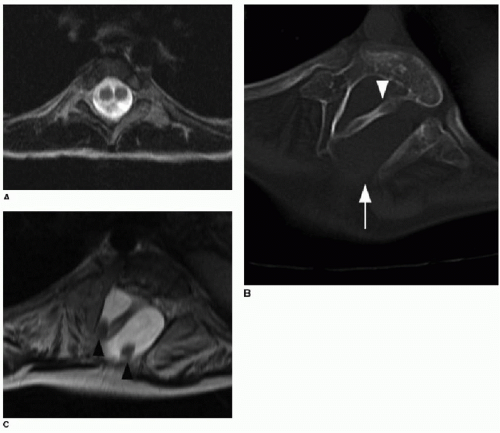 FIG. 36.7 • Examples of diastematomyelia. A: Axial T2-weighted MR image of spine in a 5-year-old child shows hemicords contained within single thecal sac and no interviewing bony spur, indicating type II diastematomyelia. B: Axial CT image of thoracic spine in different patient at age 7 years shows skewed rotation of vertebral column due to scoliosis, dysraphism (arrow), and corticated bony bar (arrowhead) dividing spinal canal into two compartments. C: Axial T2-weighted MR image of the same patient at this level shows two hemicords (black arrowheads) on separate sides of intervening bony bar, each within its own dural sac. |
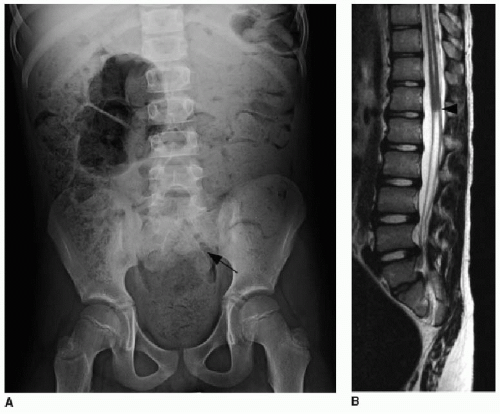 FIG. 36.8 • Caudal regression syndrome. A: Abdominal radiograph of an 8-year-old with longstanding constipation, urinary incontinence, and lower extremity spasticity shows excessive stool burden throughout colon and sacral hypoplasia (black arrow). B: Sagittal T2-weighted image of lower spine shows truncation of the distal cord (arrowhead). Sacral hypoplasia is evident as well. |
Open spinal dysraphism
Myelomeningocele accounts for greater than 98% of all OSD cases and affects 0.6 in 1,000 live births.11 In this developmental disorder, a segment of the primitive cord (the placode) fails to undergo neurulation and protrudes with meninges through a dorsal osseous defect. The placode and meninges are thereby exposed to the environment. Diagnosis may be made by prenatal ultrasound (Fig. 36.9), and if fetal surgical repair is not pursued, the myelomeningocele will be repaired soon after birth. When a myelomeningocele is present at S1 or higher, a Chiari 2 malformation will usually be present (discussed in detail in Chapter 30). Maternal dietary supplementation with folic acid during pregnancy is protective against neural tube defects and has contributed to the decreased incidence of this disease.29 A much less common type of OSD is the myelocele, or myeloschisis, which is distinguished from the myelomeningocele by location of the placode at the level of the skin, rather than displaced outward by the ballooning subarachnoid space.11
SPINE ABNORMALITIES AND LOW BACK PAIN
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


