Twenty percent of all neoplasms in children arise in the central nervous system (CNS). The incidence of CNS tumors in children has increased over the past three decades (1). The relative frequency of brain tumors by site and histology is indicated in Table 3.1. The current World Health Organization (WHO) classification of CNS neoplasms is summarized in Table 3.2 (2).
Over half of pediatric CNS tumors arise in the supratentorial brain. Anatomically, the supratentorial cranial compartment (Fig. 3.1) includes the cerebral hemispheres (frontal, parietal, temporal, and occipital lobes), the diencephalon (hypothalamus, the optic chiasm, and the thalamic, caudate nucleus, putamen, and basal ganglion structures, the latter generally considered together as the thalamic region), the pineal region (pineal gland and posterior third ventricular region), and the sellar/suprasellar region. Tumors can be categorized by anatomic region, correlating with clinical signs and specific histiotypes: suprasellar lesions (including glial tumors of the optic chiasm and adjacent hypothalamic area, craniopharyngiomas, and germ cell tumors [GCTs]), central deep-seated lesions (gliomas of the thalamic region and tumors of the pineal region, including germ cell, embryonal, and glial neoplasms), and peripheral lesions (gliomas, ependymomas, and embryonal tumors of the cerebral hemispheres).
Table 3.1 Common Brain Tumors in Children – Relative Incidence
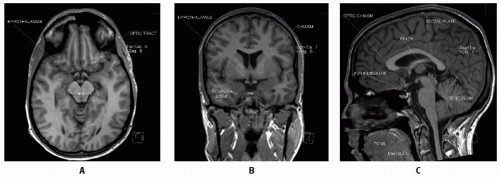
Figure 3.1 A: Axial MRI demonstrating key anatomic sites from the interorbital frontal lobes, through the superior suprasellar region (optic tracts, hypothalamus), midbrain, and superior cerebellar vermis (posterior to midbrain). B: Coronal MRI through central suprasellar location with posterior chiasm, hypothalamic nuclei, anterior aspect of third ventricle (between R and L hypothalamic nuclei), lateral ventricles (above third), and mid-temporal lobes. C: Sagittal MRI demonstrating supratentorial brain, tentorium and underlying cerebellum (with central fourth ventricle), brainstem (pons with medulla below and tegmentum of midbrain contiguously above), tectal plate (cerebral aqueduct delineates this in midline from anteroinferior tegmentum), pineal gland, and suprasellar structures (optic chiasm and infundibulum of pituitary-hypothalamic stalk).
Advances in neuroimaging have improved the accuracy of diagnosis in CNS tumors. Introduction of perfusion, diffusion, and diffusion tensor imaging may add to diagnostic capabilities. In suprasellar and pineal region lesions, computed tomography (CT) is helpful to delineate calcification typically seen in craniopharyngiomas and malignant GCTs. Positron emission tomography (PET), single photon emission tomography (SPECT), magnetic resonance spectroscopy (MRS), and magnetoencephalography are of potential value in assessing tumor type or grade, and may be helpful in differentiating tumor progression from radiation-related changes (e.g., intralesional necrosis) (3, 4, 5, 6).
Modern neurosurgical techniques have largely eliminated the need to consider multidisciplinary therapy without a histologic diagnosis (7). Central lesions within or around the third ventricle (suprasellar, thalamic, or pineal region tumors) can be biopsied endoscopically at the time of third ventriculostomy, often indicated to decompress the ventricular system in preference to a ventriculoperitoneal shunt at diagnosis. Histology may not be required for diagnosis of malignant GCTs (where elevated level of α-fetoprotein [AFP] or β-human chorionic gonadotropin [β-HCG] is diagnostic) and optic pathway tumors (OPTs) that involve the optic nerve alone or the chiasm in conjunction with adjacent optic pathway structures (8,9). For craniopharyngiomas, the imaging diagnosis can be confirmed by cyst aspiration documenting the presence of diagnostic squamous cells or cholesterol crystals in the cyst fluid (9). Both diagnostic and therapeutic benefit may be realized by second-look surgery before initial postoperative therapy or following preirradiation chemotherapy.
ETIOLOGY
Although most brain tumors are sporadic, a number of pediatric brain tumor presentations are associated with recognized neurocutaneous or other genetic syndromes, as out-lined in Table 3.3 (10).
Neurofibromatosis is a common congenital disorder associated with CNS tumors (11). Clinical criteria for type I neurofibromatosis (NF1) include six or more café au lait spots and peripheral neurofibromas among other characteristic features. NF1 is an autosomal dominant syndrome linked to a 17q11 chromosomal defect. Fully 15-20% of children with neurofibromatosis ultimately present with CNS neoplasms, usually gliomas of the optic pathways or low-grade tumors of the diencephalon, cerebral hemispheres, or posterior fossa (12). Low-grade gliomas associated with NF1 may be less aggressive than similar gliomas in the general population. As in sporadic cases, NF1 children with diffuse (infiltrating) astrocytomas (WHO grade II) do less well than those with the more common classical juvenile pilocytic astrocytomas (JPAs; WHO grade I) (13,14).
The indolent subependymal giant cell astrocytoma occurs in children with tuberous sclerosis, a hereditary disorder signified also by cutaneous acneiform lesions and hamartomatous angiofibromas in the skin, brain, heart, and kidneys, mental retardation, and renal insufficiency; CNS findings include hamartomatous periventricular lesions known as tubers. The disease is marked by the tuberous sclerosis complex gene 1 (TSC1), identified as a 9q34 mutation (15).
Childhood brain tumors (and sarcomas) are frequently noted in families with the Li-Fraumeni familial tumor syndrome; the entity is also associated with breast cancer, sarcomas, and brain tumors in adults, commonly occurring before the age of 35-40 (16). The syndrome is associated with germline TP53 mutations and a high incidence of astrocytoma (low and high grades), gliosarcoma, medulloblastoma, and primitive neuroectodermal tumor (PNET). Those with the syndrome are also at high risk for secondary, treatment-related neoplasms (17).
Table 3.3 Genetic Syndromes Related to Pediatric CNS Tumorsa
aMulvihill JJ. Clinical ecogenetics: cancer in families. N Engl J Med. 1985;312:1569-1570.
Radiation-induced meningiomas have long been recognized. Data from the Israeli experience with low-dose irradiation for tinea capitis show a striking, dose-related incidence of meningiomas (and a relatively marginal increase in gliomas), largely apparent 30+ years postexposure. Reports indicate a disturbing incidence of secondary gliomas in certain pediatric settings, with recent focus on high-grade tumors among long-term survivors of childhood acute lymphoblastic leukemia (ALL) (18,19). The estimated risk is 1-3% after routine preventive cranial irradiation (CrI); a dose-response relationship has shown a minimal incidence after dosages less than 20 Gy and a risk approximating 10% ten or more years after cumulative dosages greater than 30 Gy (19). A unique experience at St. Jude Children’s Research Hospital found a 12% incidence of malignant secondary tumors 3-5 years posttherapy in a particular study who had received 18 Gy CrI and concurrent high-dose antimetabolite therapy (6-mercaptopurine); the incidence is now over 20% in the latter trial (20).
CLINICAL PRESENTATION
Supratentorial tumors generally present with localizing neurologic symptoms; symptoms and signs may develop over extended time intervals and are often protean. Seizures are the most common symptom in cerebral hemispheric lesions, especially with tumors arising in the temporal lobe. Lateralizing neurologic signs (motor and/or sensory) occur in thalamic region tumors, often associated with symptoms of increased intracranial pressure (i.e., headaches, vomiting). Suprasellar tumors typically occlude the foramen of Monro, also resulting in symptoms of elevated intracranial pressure. Visual signs (visual field deficits and/or decreased acuity) and endocrine abnormalities (diminished growth hormone, cortisol or thyroid production; diabetes insipidus; delayed or precocious puberty) are often apparent with midline suprasellar lesions. Youngsters with suprasellar tumors may show features of the diencephalic syndrome (hyperactivity and asthenia, the latter despite normal or high food intake) (21). Pineal region tumors produce hydrocephalus by compressing the aqueduct of Sylvius; specific ocular signs (i.e., the Parinaud syndrome: decreased upward gaze, near-light dissociation of the pupillary response, convergence nystagmus) are classically noted (22).
LOW-GRADE SUPRATENTORIAL ASTROCYTOMAS
Low-grade astrocytomas are a diverse group of tumors that collectively represent the most common category of pediatric brain tumors (23). Management is variable and dependent on tumor location, patient age, presence of a genetic mutation, and, often, physician and parental preference. Outcomes are generally favorable; the goal of treatment is durable disease control or cure with preservation of function. The most frequent site of origin is in the cerebellum (see Chapter 4) followed by the deep midline diencephalon (thalamus, hypothalamus, optic chiasm and nerves, basal ganglia, and related structures) and the cerebral hemispheres. The most common locations for the hemispheric tumors are the frontal and temporal lobes (24, 25, 26, 27). In contrast to adults who are most commonly diagnosed with high-grade astrocytomas, the majority of supratentorial tumors occurring in childhood are low-grade astrocytomas. Low-grade gliomas of the optic pathway and hypothalamus have distinct features and are discussed below as a separate group.
A subset of diffuse low-grade tumors extend across two or three lobes often with no obvious or dominant mass lesion; the diffuse pattern of involvement is classified as gliomatosis cerebri (28, 29, 30). Typical unifocal low-grade gliomas occasionally present with subarachnoid dissemination; the incidence is approximately 3%, most often seen in tumors located in the diencephalon (31,32).
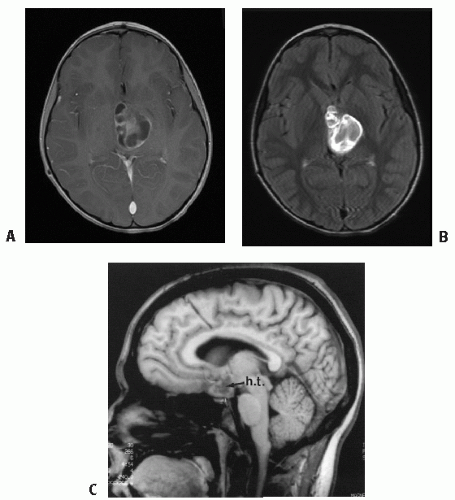
Figure 3.2 A: JPA of (L) thalamus, presenting in a 4-year-old boy—cystic and solid, largely enhancing tumor on T1-gadolinium MRI. B: JPA visualized on T2-FLAIR sequence post-contrast. Note even better tumor definition. C: Note leptomeningeal metastasis in hypothalamus/chiasmatic JPA, a finding largely at diagnosis in <10% of cases.
Diagnostic imaging generally reveals a lesion that is isointense on CT and T1 MRI sequences and hyperintense on T2 sequence (33). JPAs enhance briskly and diffusely, but most other low-grade tumors show little or no enhancement with gadolinium (33, 34, 35). Scattered calcifications may be present. Tumors may include small or dominant cysts; some degree of cyst formation is common in JPAs. MRI is the imaging modality of choice. Due to the variability of enhancement, T1 sequences may not provide the best demarcation of the tumor borders; post-gadolinium T2-FLAIR images are often preferred (Fig. 3.2B).
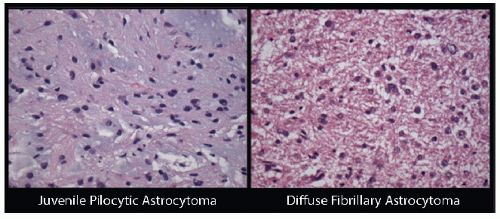
Histopathology of low-grade astrocytomas is characterized by low cellularity, little nuclear atypia, and few, if any, mitotic figures. Although the term “low grade” applies to all pediatric gliomas that are not anaplastic, the various histiotypes differ in the degree of infiltration, relative aggressiveness, and prognosis (36). JPAs and diffuse fibrillary astrocytomas comprise the majority of pediatric low-grade gliomas. Figure 3.3 depicts histological features of these two most commonly encountered low-grade astrocytomas. Less common low-grade astrocytomas include gemistocytic astrocytomas, pleomorphic xanthoastrocytomas, desmoplastic infantile astrocytomas, protoplasmic astrocytomas, and subependymal giant cell astrocytomas. These tumors are generally classified by the WHO histological criteria (2,37). The WHO system categorizes JPA as grade I and other differentiated astrocytomas including all diffuse astrocytomas, as grade II; anaplastic astrocytoma and glioblastoma multiforme are coded as grade III and IV, respectively (2,37). Broniscer et al. reported a cumulative rate of malignant transformation amongst pediatric grade II astrocytomas of 7% at 15 years post-diagnosis, a much lower frequency than has been reported in adults (38). JPAs almost uniformly retain low-grade characteristics even when uncontrolled or recurrent (38).
Figure 3.3 Histological features of the most common pediatric low-grade gliomas: JPA (L) and diffuse fibrillary astrocytoma (R). JPAs (WHO grade I) show biphasic architecture with more cellular areas that tend to have the Rosenthal fibers and loose areas with microcysts. Cells are elongated, bilpolar, and have thin “hairlike” processes. Diffuse fibrillary astrocytomas (WHO grade II) consist of long, thin cells highlighted by a crisscrossing background of cytoplasmic glial filaments; nuclear atypia often apparent.
JPAs are characterized by elongated bipolar astrocytes organized in a parallel array of glial fibers, giving a hairlike or piloid appearance (39). The sparsely cellular tumors are signified by microcysts and Rosenthal fibers (amorphous eosinophilic material formed by plump, degenerating astrocytes). Macroscopically, visible cystic components often are present, sometimes with less prominent solid tumor components. The tumors may be circumscribed or may extend along white matter tracts, depending on the location (Fig. 3.4) (39). JPA is the most common tumor in the diencephalic region and comprises 25% of hemispheric astrocytomas (7,25). The tumor typically is nonaggressive, although multifocal presentation, rapid progression, and dissemination may occur (31,40, 41, 42).
Pilomyxoid tumors have recently been defined as a subset of low-grade gliomas, previously classified with pilocytic astrocytomas (43,44). Pilomyxoid tumors have a myxoid matrix with highly monomorphic piloid cells; they may demonstrate an arrangement resembling perivascular rosettes; they are usually solid with less prominent cystic components and lack Rosenthal fibers (45). These tumors are felt to be more aggressive than classical pilocytic astrocytomas and are categorized by the WHO as grade II, although little data yet confirm the clinical implications of this diagnosis (2,44).
Grade II astrocytomas are characterized by a greater degree of infiltration; the proliferation index may exceed that of the JPA, and a greater number of molecular aberrations are apparent. Diffuse fibrillary astrocytomas consist of long, thin cells highlighted by a crisscrossing background matrix of cytoplasmic glial filaments. Gemistocytic astrocytomas are less common, presenting as benign-appearing, often circumscribed lesions with a high rate of recurrence; these tumors often demonstrate malignant degeneration within several years after initial presentation (46). Protoplasmic astrocytomas are characterized by large, rounded astrocytes with abundant cytoplasm and a background largely devoid of fibrils (47). Grade II astrocytomas are more often associated with local recurrence and may present with malignant progression (38,48,49).
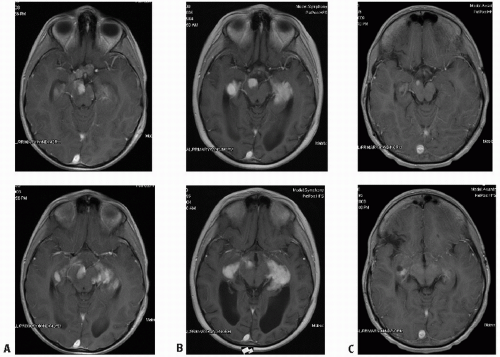
Figure 3.4 A: A 7-year-old boy with JPA of optic chiasm and optic tracts (without neurofibromatosis), involving and distorting chiasm (A, top) with spread along optic tracts (A, bottom). B: Increased tumor size, enhancement along optic tracts noted 7 months postradiotherapy (post-RT), attributed to transient posttherapy effects (B, top and bottom). C: Same levels with marked, spontaneous reduction in tumor volume compared to pre-irradiation or MRI 7 months post-RT (Figures B); note normal chiasm (C, top). Patient now 5.5 years post-RT, with intervening (R) middle cerebral infarct (C, bottom; same time as C, top).
Pleomorphic xanthoastrocytomas present as superficial tumors in the cerebral hemispheres. Despite an aggressive histological appearance with cellular pleomorphism and numerous mitoses, the behavior of pleomorphic xanthoastrocytomas is quite benign; the tumor is classified as grade II. Long-term disease control usually is obtained through surgery alone; a proportion of cases do recur late and occasionally show malignant degeneration (50,51). A small percentage of these lesions show mitotic activity, areas of necrosis, and a high proliferative index at initial diagnosis and are designated as anaplastic pleomorphic xanthoastrocytoma. The latter may be somewhat more aggressive clinically (52,53).
Subependymal giant cell astrocytomas are a distinct subtype of sharply marginated tumors that occur along the linings of the lateral ventricles (2). The lesions occur in the setting of tuberous sclerosis (v.s.); children often have seizures and show significant developmental delays (54). Subependymal giant cell tumors are almost always managed with surgical resection alone.
Therapy
Surgery
For low-grade gliomas amenable to complete surgical resection, surgery is generally the first and sole intervention providing excellent control of disease. Total resection is usually feasible for tumors arising in the cerebral hemispheres with reports indicating gross total resection (GTR) in 90% of cerebral hemispheric astrocytomas (25,26,55). Low recurrence rates are reported for both JPA and other astrocytomas after total resection (25,26,55, 56, 57). Data from the Children’s Cancer Group (CCG) and Pediatric Oncology Group (POG) study of primary surgery in low-grade gliomas indicate 5-year progression-free survival in 92% of children after GTR, ranging from 95-100% for JPAs and gangliogliomas to 80% for grade II diffuse astrocytomas in this age group (58). Resection of tumors involving the dominant medial temporal lobe, motor strip region, or the Broca speech cortex may not be possible without inducing severe neurologic deficits. Partial resection may provide initial intervention for decompression and histopathologic diagnosis.
Low-grade gliomas of the diencephalon are technically challenging due to the deep location and eloquent area; however, contemporary series report successful resection for selected tumors in this region (59, 60, 61, 62, 63, 64, 65, 66). Long-term disease control is typically achieved by total resection; occasional regression of diencephalic JPA after partial resection alone has been reported (59,65,66). The CCG-POG study notes a progression-free survival after surgery alone of 50-60% for diencephalic tumors, much lower than for lesions originating in the cerebellum or cerebral hemispheres where GTR is more common (58).
Radiation Therapy
Radiation therapy is an established, effective treatment for low-grade gliomas, achieving tumor response and durable disease control in a significant proportion of pediatric cases (Fig. 3.2) (26,56,67, 68, 69, 70). An analysis from Pollack et al. showed improved disease control at 10 years after irradiation following incomplete resection of cerebral hemispheric astrocytomas: 82% progression-free survival with irradiation versus 42% after surgery alone. The same study showed no significant benefit in overall survival (26). A recent phase II prospective study published by Merchant et al. demonstrated excellent event-free survival (87% and 74% at 5 and 10 years, respectively) and overall survival over 90% at 10 years for children treated with three-dimensional (3D) conformal irradiation to the MRI-defined tumor volume (71). These outcomes compared favorably to the literature and have been associated with excellent functional outcomes (72). The decreased volume of normal brain exposed to moderate or high radiation doses using conformal techniques with small margins may significantly decrease some of the serious radiation-induced side effects (72,73). Care must be taken to accurately delineate tumor volumes, addressing white matter tracts in defining the clinical tumor volume (CTV). Merchant et al. suggest a minimal margin (as small as 5 mm) may be adequate for well-demarcated tumors (71). Children’s Oncology Group (COG) is currently examining outcomes with the use of a 5-mm anatomically limited CTV margin around the gross tumor volume (GTV) in a multi-institutional trial.
There are no contemporary data suggesting a benefit to postoperative irradiation for completely resected low-grade astrocytomas. For incompletely resected low-grade astrocytomas, early administration of irradiation may not benefit the patient (74). Current indications for radiation therapy after a near total resection (with imaging evidence of disease residual) include symptoms or signs that might improve with irradiation or postsurgical progression in a location not amenable to safe, definitive second resection. Other factors that are taken into consideration are histological subtype or biology (57,75). Chemotherapy may be advised prior to irradiation for young children or for patients with neurofibromatosis (NF1) or cognitive delay.
Reports from the large CCG-POG low-grade trial confirm previous institutional data suggesting that a significant proportion of incompletely resected astrocytomas remain indolent over 3-5 years; progression-free survival rate at 5 years is 55% after near total or subtotal resection (58). A decision to observe children with residual astrocytoma should involve all subspecialties (neurosurgery, radiation oncology, pediatric oncology) indicating the obligation for regular clinical follow-up and imaging that allows one to comfortably observe a child with the anticipation that treatment can be initiated upon documented tumor progression (55). Such an approach balances the recognized efficacy of radiation therapy with potential toxicities related, in part, to the anatomic location and volume of the tumor and the age of the patient (13,27). Inherent in such an approach is the commitment to intervene appropriately with documented disease progression, including use of primary radiation therapy, additional surgery, or chemotherapy as indicated.
JPAs predominate in hypothalamic and OPTs. Prolonged progression-free survival after irradiation alone for these tumors reflects both the indolent nature of tumors in this site and the efficacy of irradiation. Survival rates in excess of 80% at 10 years after irradiation are common for these tumors (9,73). In thalamic astrocytomas, pilocytic histology is less prevalent; 10-year survival results range from 33% to 60% after therapy (9,71,73,74,76).
Anatomically, diffuse low-grade gliomas are uncommon outside the brainstem in children. Gliomatosis cerebri or bithalamic astrocytomas may represent supratentorial counterparts of the more common infiltrating pontine astrocytomas (76,77). These tumors respond to radiation therapy both symptomatically and by imaging, although recurrence and progression often are apparent within a year (76,77).
Chemotherapy
Chemotherapy has been used with increasing frequency for low-grade gliomas as a strategy to delay or avoid radiation therapy; less data is available regarding chemotherapy response for progressive disease following irradiation. The use of initial chemotherapy for low-grade gliomas has been studied primarily in centrally located tumors that tend to occur in younger patients and are not amenable to resection. Chemotherapy can provide disease control for months to years, more often achieving stable disease or partial response than complete resolution; most tumors progress within 3-4 years, requiring radiation therapy at that time. The age below which chemotherapy is more appropriate is controversial and dependent on factors such as tumor size and location, presence of NF mutation, or developmental or neurocognitive delays. Packer et al. reported age to be the only significant prognostic factor, with 3-year progression-free survival rate of 74% for children ≤5 years old versus 39% for older children (78). In COG, the age of ≤10 years has been chosen for trial eligibility in studies evaluating chemotherapy as initial treatment (79). Individual centers have used thresholds of <3 or <5 years old.
Favorable control rates and relative absence of serious toxicity have established carboplatin and vincristine as the “standard” first-line chemotherapy for low-grade gliomas in younger children (78,80, 81, 82). Carboplatin hypersensitivity occurs over time, often limiting further administration of this regimen (83). Temozolomide, an alkylating agent with modest responsiveness in recurrent low-grade gliomas, is currently in trial with carboplatin/vincristine to try to prolong drug tolerance (84, 85, 86, 87). The five-drug University of California at San Francisco (UCSF) regimen (6-thioguanine, procarbazine, dibromodulcitol, lomustine, and vincristine) has been reported to be similarly efficacious; a randomized trial comparing carboplatin/vincristine to this regimen has been completed in COG, suggesting equal or greater efficacy with the five-drug regimen (79,88). Bevacizumab in combination with irinotecan has been investigated for recurrent low-grade glioma with promising response rates for heavily pretreated patients (89). The decision to proceed with chemotherapy or irradiation relates to patient age and clinical presentation; consideration should involve consultation with and discussion among a multidisciplinary team reviewing relative risks and benefits with the patient’s family. Factors beyond age alone include symptoms and signs, potential for additional neurocognitive deficits, the likelihood of durable benefit from respective chemotherapeutic regimens, and the radiation volume required in weighing relative potential toxicities. Although not proven to be problematic, there is no confirmation that the response rate and durability of disease control following radiation therapy are independent of prior failure on chemotherapy.
Radiotherapeutic Management
Volume
Fusion of MRI with CT imaging is ideal to determine the extent of disease defining the GTV for treatment planning. T1 gadolinium-enhanced imaging may be ideal for JPA; for diffuse astrocytoma (grade II) and the less common low-grade neoplasms, T2-FLAIR or T2 sequences are most useful where the tumor rarely shows contrast enhancement throughout. The CTV may be defined as a 1-cm anatomic expansion; a prospective study at St. Jude indicated a dearth of marginal recurrences with a 1-cm anatomic expansion of the GTV to define the CTV (70,71). Smaller margins may provide adequate control; a 5-mm anatomic expansion is being examined in a prospective COG study. An additional 3-5 mm geometric expansion defines the planning target volume (PTV). The use of small margins requires a careful review of all neuroimaging with great attention to detail including all cystic and solid components of the tumor in the GTV. Similar guidelines appear to be appropriate for grade II (fibrillary) astrocytomas, recognizing that most such tumors in children have readily identifiable margins on imaging; expansion of the GTV by 1 cm has resulted in few marginal failures (70,90). Shrinking fields typically are not used in JPAs; when target volumes for infiltrating fibrillary astrocytomas are used, fields may be reduced to more narrowly encompass the obvious tumor after delivery of 50.4 Gy. For tumors with a cystic component, a CT or limited sequence MRI during radiation therapy may prove useful to evaluate changes in the cystic portion of the tumor during treatment. Diffuse low-grade gliomas can be associated with gliomatosis cerebri or bithalamic astrocytomas; both may be histologically WHO grade II, but the pattern of growth indicates a degree of aggressiveness and infiltration requiring large volume irradiation based on guidelines for malignant gliomas (76,77). For gliomatosis cerebri that is bilateral and involving much of the cerebral hemispheres, corpus callosum, and, sometimes, thalamic nuclei, it may be appropriate to include the whole brain, at least to a dosage level of 40-45 Gy before using a more imaging-defined “boost” volume (77,91).
Astrocytomas with multifocal presentations or subarachnoid seeding generally are treated with craniospinal irradiation (CSI), although the need to cover the entire neuraxis is controversial and unproven (31,40,41).
Dosage
There is little definitive dose-response data specific for pediatric astrocytomas. Shaw et al. showed an advantage with radiation therapy in JPA and other low-grade astrocytomas only at doses of greater than 53 Gy (91). It seems ironic that the adult low-grade glioma trials have established no clear benefit for dosage levels greater than 45 Gy (the European Organization for Research and Treatment of Cancer study showed no difference between 45 and 59.4 Gy; the North Central Cancer Treatment Group, Radiation Therapy Oncology Group, and Eastern Cooperative Oncology Group study showed no difference between 50.4 and 64.8 Gy) (92,93). Pediatric national trials continue to use doses approaching the established tolerance of critical CNS structures (i.e., 50-54 Gy). The primary justification is the long-term disease control rates documented in studies of pediatric JPA and other low-grade astrocytomas. For children younger than 5 years, a delay in irradiation is appropriate as clinical signs permit, particularly with increasing data indicating the efficacy of chemotherapy in delaying irradiation for an average of 3-5 years. When radiation therapy is needed, a dose of 50-54 Gy with optimal conformality is standard-the same dose level recommended throughout the pediatric age range (70,71).
Technique
Ideal techniques achieve close conformation of the high-dose region to a well-defined local target volume. The 3D conformal or intensity-modulated photon irradiation or proton beam radiation therapy can achieve such conformality, with potential differences in sparing adjacent dose-limiting structures (e.g., optic chiasm, eye, and pituitary-hypothalamic region) and in intralesional homogeneity. Given the nature of low-grade gliomas and recognized dose-related toxicities, attention to dose volume histograms (DVH) and a 3D image-based radiation plan is critical in designing treatment techniques. Multiple fields, based on single-plane or non-coplanar techniques, may be used to achieve a dose distribution maximally conformed to the target volume (Fig. 3.5). The impact of low-dose irradiation on broader regions of the developing brain has not yet been fully explored; dose-volume modeling suggests correlations between low dose volumes and cognitive deficits (94). Early results suggest that 3D conformal techniques achieve greater sparing of neuropsychological function, for example, than past experience has shown for conventional therapies (95). The physical properties of protons allow for further reduction in the amount of brain exposed to low-dose irradiation (96,97) (Fig. 3.6). The greater conformality of 3D conformal radiation therapy, intensity-modulated radiotherapy (IMRT), and proton therapy are likely to be associated with less pronounced late toxicities as a result of diminished high-dose exposure of normal tissues (96,98, 99, 100, 101, 102).
The use of single-fraction radiosurgery for primary management of pediatric low-grade gliomas has been reported in several small institutional series (103,104). There are less data to understand the potential risks and benefits of this approach; however, in the rare instances where small lesions distant from critical structures can be targeted, especially in children with NF1 where subsequent development of additional CNS tumors is relatively common, data suggest reasonable efficacy and intermediate-term tolerance (103, 104, 105).
Results
Overall, outcomes for low-grade gliomas are quite favorable. Prognosis is related to age at diagnosis and degree of surgical resection; older children usually enjoy more favorable outcome. GTR is consistently linked to improved progression-free survival (26,58,106, 107, 108, 109, 110). Although low-grade gliomas are often considered one entity for the purposes of clinical trials and management, they represent diverse histiotypes with variable prognoses as documented in series reporting detailed histopathologic analyses (36,111). Advances in pathology, particularly better molecular and genetic classification, should further categorize tumors and lead to the development of targeted agents and modifications in treatment.
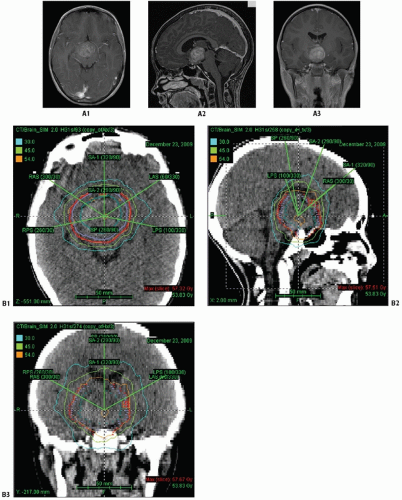
Figure 3.5 Optic chiasm/hypothalamic glioma presenting for radiation therapy at 4 years old following imaging evidence of progression after two chemotherapy regimens (A1-A3) B: target volumes for this case depicted for axial (B1), sagittal (B2), and coronal (B3) planes. Note CT dosimetry using IMRT 6 MeV photons with: GTV (green) with 5-10 mm anatomic expansion for CTV (blue); geometric 3-mm expansion for PTV (red).
Long-term disease-free survival has been reported in 90% of children after complete resection of cerebral hemispheric tumors; for similarly managed thalamic tumors, smaller series report 60-90% survival (26,55,65,67,112). Incomplete resection alone results in approximately 50% progression-free survival at 5 years (58). The addition of therapeutic irradiation has been shown to achieve progression-free survival in hemispheric astrocytomas in approximately 80% of children measured at 10 years (26,71); indications for therapeutic (rather than routine adjuvant) irradiation are discussed earlier in this chapter. For thalamic tumors treated primarily with irradiation, 40-50% survive free of progression (7,9,64,76). Hypothalamic tumors have a favorable outcome after primary irradiation, with 10-year progression-free survival rates of 70-85%; current literature typically groups these tumors with chiasmatic lesions as “hypothalamic/chiasmatic gliomas” as discussed below (7,9,68,113). Particularly when one is treating central (diencephalic) JPAs, it is noteworthy that tumor size may appear to increase approximately 6-12 months after irradiation, often by apparent intralesional necrosis or cyst formation; such changes typically remit over several months. The majority of lesions respond by imaging criteria after radiation therapy (both by size and gradual loss of enhancement), but the median time to objective response is greater than 15 months (Fig. 3.4) (68,114). Low-grade gliomas involving both right and left thalamic regions (i.e., bithalamic tumors) have an aggressive course similar to that of the more diffuse gliomatosis cerebri or pontine gliomas (76).
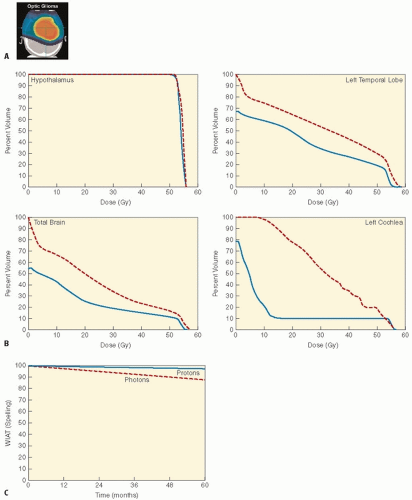
Figure 3.6 A: Radiation dosimetry for optic chiasm glioma using scanning proton beam. B: DVH derived as a composite plan from 10 cases of optic chiasm gliomas, comparing 3D conformal RT (photon is red/dashed) and scanning proton beam (blue/solid). C: Estimated IQ change based upon dose-volume effect models, depicting Wechslar Individual Achievement Test (WIAT) spelling for 3D conformal RT (photon, red/dotted) and scanning protons (blue/solid). (From Merchant TE, Hua CH, Shukla H, et al. Proton versus photon radiotherapy for common pediatric brain tumors: comparison of models of dose characteristics and their relationship to cognitive function. Pediatr Blood Cancer. 2008;51: 110-117, with permission.)
OPTIC PATHWAY TUMORS
OPTs represent 5% of childhood brain tumors. OPTs may involve one or several anatomic sections of the optic system (optic nerves, optic chiasm, optic radiations) (Fig. 3.4) (13,68,115, 116). They can be small and localized or extensive and infiltrative. Tumors involving the chiasm are often difficult to distinguish from tumors originating in the hypothalamus; hence, chiasmatic gliomas are commonly grouped with hypothalamic gliomas as one entity (113). OPTs occur predominantly in young children; 25% present before 18 months of age, and 50% present before 5 years (68,113,115,116). Up to 25% of childhood OPTs occur in children with NF1 (61,68,81). Conversely, of children with NF1, 10-20% are found to have OPTs on MRI; careful neuro-ophthalmological examination ± screening MRIs should be standard for patients with NF1 (61,68,81,113, 117,118).
Clinical presentation is most often with diminished vision. In young children, increased intracranial pressure endocrinopathies and diencephalic syndrome may predominate. Histologically, more than 90% of OPTs are low-grade astrocytomas, most often JPAs (more than 65% of all cases) and grade II astrocytomas (25%), with infrequent gangliogliomas or hamartomas; malignant gliomas are uncommon (13,63,69,113,119).
The nature of OPTs has been debated. While acknowledging the sometimes indolent nature of OPTs, serial observations in major pediatric neuro-oncology clinics indicate progression in 75-85% of children, typically within 2 years of initial presentation (8,13,61,113,115,116). Tumors confined to the optic nerve(s) may behave in a more hamartomatous fashion. Children with NF1 have a more indolent course with lower rates of progression and longer latency intervals (113,118). Other signs of the neoplastic behavior of OPTs include extension to or invasion of the adjacent hypothalamus and posterior extension to the optic tracts (up to and including the lateral geniculate bodies) and optic radiations. Infrequently, optic chiasmatic and hypothalamic tumors demonstrate diffuse leptomeningeal disease (31,32,40, 41, 42). Mortality within 10 years of diagnosis is uncommon, although ultimate disease-related mortality has been documented in up to 40% of cases (9,13,68,115, 120,121). Also to be noted is the rare but documented occurrence of spontaneous regression of OPTs (122,123).
Several series identify chiasmatic OPTs as optic pathway/hypothalamic gliomas, acknowledging the difficulty in identifying the origin of tumors that intimately involve both the chiasm and the hypothalamus (81,124). Although lesions extending to or originating in the hypothalamus may be somewhat more aggressive than lesions confined to the visual pathways, up to 50% of selected, asymptomatic children have been free of progression for 5 years or longer without therapeutic intervention. Preliminary data suggest adequate retrieval with secondary therapy at the time of progression during observation (13,113,121).
Therapy
Surgery
Surgery has been the preferred treatment for unilateral tumors of the optic nerve (115,116,119). Observation may be selected, especially if there is residual vision associated with a lesion confined to the intraorbital optic nerve. Alvord and Lofton (115) reported progression in 70% of children with untreated lesions within 6 years of diagnosis, although it was rarely associated with tumor-related mortality. Most series indicate more indolent, perhaps truly hamartomatous behavior in children with neurofibromatosis (119,121).
For lesions involving the optic chiasm, there are limited data suggesting a role for surgical resection. Decompression or limited resection may be successful in restoring vision (62,63,125). Series from the Hospital for Sick Children, Toronto, and New York University suggest a somewhat broader role for local excision in selected presentations. The latter authors indicate removal of lesions of partially infiltrating low-grade optic pathway astrocytomas with surprisingly little added visual compromise; approximately 50% of such cases have remained stable without further intervention for 3-5 years (60,63,119,125). Whether less aggressive surgery or observation would have achieved similar results relates to the selection of cases for surgery, a debate in the surgical community (8). Care should be taken to avoid visual compromise or other surgical complications as alternative therapies are quite successful.
Typical chiasmatic lesions that involve components of the visual pathways beyond the optic chiasm and hypothalamic region (i.e., with imaging extension to the optic nerves, optic tracts, or optic radiation) may be managed without biopsy confirmation. Most of these tumors are low-grade astrocytomas and most often can be managed based on the clinical and imaging diagnosis. Globular tumors that involve the chiasm and hypothalamus are best biopsied if this can be performed safely; a small percentage of these lesions may be GCTs, unusual types of low-grade neoplasms, or more aggressive malignant gliomas (62,69).
Radiation Therapy
Irradiation is indicated for significant visual or neurologic deficits at presentation or documented progression by clinical evaluation or neuroimaging after observation, surgery, or chemotherapy (13,61,78,81,82,115). Radiation therapy is highly effective for OPTs: 10-year progression-free survival rates exceed 80% (13,68,69,126). Although overall survival at 10 years is unaffected by the initial therapeutic approach (i.e., observation, resection, chemotherapy, or irradiation), progression-free survival rates at 5 and 10 years are substantially higher after radiation therapy (9,13,55). Serial imaging studies document significant tumor response in more than 50% of children after irradiation (69,113). Transient postirradiation tumor enlargement, often in the setting of central cystic degeneration, has been well documented (68). Close observation and medical management (i.e., steroids) rather than aggressive intervention for presumed tumor progression is advised, particularly for lesions that may appear to progress within 6-12 months after radiation (68). Visual improvement has been reported in 25-35% of children after irradiation (68,113,126). Visual deterioration is reported in 10-20% of children after irradiation, largely related to cystic degeneration (and consequent increased mass effect at the chiasm) or unrecognized elevated intracranial pressure (13,68,113,126). Vision should be monitored closely during the radiation course and in the months following completion.
Balancing the indications for radiation therapy and the timing requires judgment and, often, serial evaluations to determine the nature of the disease process. OPTs are associated with unique late radiation-related sequelae. The young age at diagnosis, central location, and often extensive anatomic involvement challenge the ability to deliver adequate radiation therapy while preserving neurocognitive function; the problem is further accentuated in children with NF1, itself associated with cognitive delays (69,81,113,118). There is also concern about late vascular events: the incidence of occlusive vasculopathy at the circle of Willis in children with brain tumors is highest among those with OPTs, especially in younger children (69,81,127,128). Moyamoya syndrome is characterized by obliteration of the major vessels at the circle of Willis; incomplete brain perfusion is provided by collaterals and peripheral meningeal vessels. The syndrome has been noted after irradiation, perhaps related to cicatricial constriction of vessels tracking through chiasmal and hypothalamic gliomas. An incidence approaching 18% has been reported, especially noted among children less than 2-3 years and those with NF1 (69,127). A baseline MRA prior to irradiation to document vascular status is important, often revealing occult vascular compromise due to the tumor itself; the baseline is important to more easily detect changes after treatment. There is also a heightened concern regarding the risk of second malignancy in patients with NF1 (129,130). The Toronto group has uniquely reported a 10% incidence of second malignant neoplasms after irradiation for OPTs; of interest, a series from Children’s Hospital of Los Angeles showed the same rate of anaplastic degeneration in JPA after surgery alone (112, 121,131).
Chemotherapy
Because of the radiation-associated morbidities in young children with OPT, Packer et al. (132) explored primary chemotherapy in children younger than 5 years. Initial experience with actinomycin D and vincristine resulted in stabilization in a majority of children and objective tumor reduction in approximately 25%. Although more than 60% of children needed irradiation by 5 years after diagnosis, the approach resulted in a substantial delay in radiation therapy, with a median time to progression of 3 years (132). Subsequent experience with an 18-month regimen of carboplatin and vincristine for low-grade gliomas including those of the hypothalamic region and OPTs has shown a significant rate of objective tumor reduction (reporting greater than 50% tumor reduction in one third of patients), early progression in only 10%, and 3-year progression-free survival that ranges from 75% for children younger than 5 years to 39% for those older than 5 years (78,81). Similar results have been reported with the UCSF five-drug regimen (88). Children with low-grade gliomas of this location are included on the recent and current COG studies discussed earlier in this chapter.
Early experience suggests favorable outcome with secondary irradiation after progression during or after chemotherapy; recent observations related to the timing of initiating radiation therapy suggest that long-term disease control and function are not diminished with prolonged preirradiation intervals (13,78,113). Toxicity with carboplatin and vincristine has been limited, and early data suggest continued intellectual development during chemotherapy (81). There is a balance between duration of disease control (clearly superior with radiation therapy) and less durable control with chemotherapy apparently without the serious morbidities associated with irradiation in the younger age group (13,113,124). In current practice, most children below 5-10 years receive chemotherapy as the initial intervention, with some centers extending this to all prepubertal children. It is important not to avoid irradiation even in younger children when progressive visual loss is apparent despite chemotherapy.
Radiotherapeutic Management
Volume and Technique
The volume of disease can be quite variable for OPTs. The target volume includes only the radiographically involved aspects of the optic pathway. MRI has enhanced the ability to better define segments of the optic pathway that are involved. Tumors are often confined to the suprasellar region for lesions that are limited to the chiasm with or without hypothalamic involvement. Central lesions necessitate multifield, often noncoplanar arrangements to optimize 3D conformal approaches, limiting the dosage to the surrounding cerebral hemispheres and minimizing intralesional inhomogeneity of dose (71,100). Proton radiation therapy provides a dosimetric benefit for such tumors (101). Optic nerve involvement, most often in NF1, necessitates inclusion of the nerve to the posterior aspect of the globe in defining the target volume. For lesions extending along the optic tracts or beyond (occasionally to involve the optic radiation, toward or to the occipital lobes), it is key to discern where apparent neoplastic changes differ from edema (68,118).
Dosage
The extensive literature on radiation therapy for OPTs typically calls for dosage levels of 50-54 Gy, most often at 180 cGy per fraction (68,116,121,126). Reduction to 50 Gy at 150 cGy daily may be appropriate in children younger than 3 years (9,126).
OLIGODENDROGLIOMA
Oligodendrogliomas represent 1-2% of supratentorial tumors in children (133). The generally circumscribed tumors occur most often in the cerebral hemispheres. In adults, loss of chromosome 1p or 19q is noted in 50-80% of cases and is predictive of response to treatment (134,135). Specific data for pediatric oligodendrogliomas are limited but suggest that similar molecular findings are uncommon in adolescents and not apparent in children younger than 10 years (30,136,137). Treatment recommendations are based largely on adult experience with surgery and irradiation (133). Adults show excellent response to procarbazine, lomustine, and vincristine (PCV) or to temozolomide chemotherapy, particularly in anaplastic oligodendrogliomas with isochromosome 1p or p53 mutations (138, 139, 140). Given differences in biology, it is unclear whether chemosensitivity can be extrapolated to children.
Total surgical resection is the treatment of choice for accessible lesions. GTR has been documented in 20-25% of all cases, apparently more often in children and adolescents (133,141). The survival rate at 10 years after total excision is reported to be 60% in the Mayo Clinic series; of six children, five survived (91,141).
For incompletely resected oligodendrogliomas, a short-term benefit for radiation therapy has been documented. Shaw et al. (91) reported 5-year survival rate of 25% after subtotal resection, compared with 62% with the addition of irradiation to doses greater than 50 Gy; by 10 years, the survival rates were 31% with irradiation and 25% without irradiation. Adjuvant irradiation typically is withheld for differentiated oligodendrogliomas in children, even with incomplete resection. Histologic grade has been cited as a prognostic indicator in oligodendrogliomas (91,142). Anaplastic oligodendrogliomas are managed similarly to other malignant supratentorial gliomas in children, although the outcome tends to be superior to those with anaplastic astrocytoma and glioblastoma (143,144).
Combined radiation therapy and PCV has been associated with excellent disease control in adults, specifically with anaplastic oligodendroglioma (145). More recently, response to Temodar has also been demonstrated in adults (134). Limited chemotherapy has been associated with sufficient tumor reduction to permit delayed GTR in tumors initially felt to be unresectable (146).
The “benign” nature of oligodendrogliomas is open to some question, with few reports documenting survival rates greater than 25% beyond 15 years (91). Long-term reports after contemporary surgery and irradiation are limited in the literature; given the added facility of stereotactic neurosurgery, one might anticipate improved outcome in the larger proportion of children with grossly resected lesions (91).
GANGLIOGLIOMA
Gangliogliomas are uncommon, biologically benign neoplasms comprised of neuronal and glial elements (ganglion cells and astrocytes, respectively) (2). The tumors occur primarily in children and young adults, with a median age approximating 25 years (149). Gangliogliomas present most often in the mesial aspect of the temporal lobes, with seizures as the dominant symptom (2,148,149). Pediatric tumors uncommonly present in the posterior fossa. The lesions are typically well circumscribed and surgically resectable (148, 149, 150). Gangliogliomas are classically coded as WHO grade I, well differentiated histologically with no atypia or anaplasia (2,149). Surgery alone is the standard initial intervention; 10-year disease-free survival has been reported in 97% of pediatric cases after surgery (147,149). Malignant transformation at progression or recurrence is rare; at most 10% of cases show nuclear atypia or anaplastic components (grade II or III, respectively) (150, 151, 152). Malignant degeneration to glioblastoma (grade IV) is decidedly uncommon in children and adolescents (151,152). Prolonged progression-free survival has been noted in small series with irradiation following incomplete resection or recurrence; the efficacy following malignant degeneration is less apparent (151, 152, 153).
RARE LOW-GRADE NEOPLASMS
Neurocytomas are clinically indolent tumors that present as intraventricular lesions, usually in the lateral ventricles with attachment to the midline septum pellucidum; most are diagnosed in adolescents and young adults. Neurocytomas are composed of small neuronal cells thought to represent a benign neoplasm derived from cells midway in the maturation process of neuronal differentiation (147,148). These tumors are genetically distinct from the oligodendrogliomas and dysembryoplastic neuroepithelial tumors (DNETs), with which they can be confused both clinically and histologically (149). The lesion generally is resectable (150). Some consider hemispheric neurocytomas more likely to be DNETs; the lesions seem to necessitate only local excision (151). Prognosis has been related to the rate of proliferation (gauged by MIB-1) (150). These tumors respond to irradiation; small series have suggested improved outcome in cases with less than total resection when followed by radiation therapy (local target volume; dose >54 Gy in 30 fractions reported to be superior to lower doses) (147,152, 153, 154).
DNETs are biologically indolent, often large cerebral cortical tumors typically presenting with a long-standing seizure history (145,155). Symptoms typically arise in children younger than 12 years; the mean age at diagnosis is 14 years (156). The tumors may be considered quasi-hamartomatous, classically are well demarcated, and show no contrast on MRI; uncommonly, DNETs present as complex solid and cystic lesions with enhancement, calcification, and intralesional hemorrhage (2,156,157). These tumors may be followed, but surgery often is needed for seizure control; although they appear to be responsive to irradiation, there is no documented role for postoperative therapy (145,155,158).
MALIGNANT GLIOMAS
Supratentorial malignant gliomas represent approximately 6% of brain tumors in children. Histologic grading divides high-grade (or malignant) gliomas into anaplastic astrocytoma (WHO Grade III) and glioblastoma (WHO Grade IV) (2,159). Children have a higher proportion of anaplastic astrocytomas among the malignant gliomas and have arguably longer survival intervals (2,160,161). The tumors are biologically separate from the more common adult malignant gliomas (162,163). Adult primary malignant gliomas appear to arise de novo and are associated with amplification of the epidermal growth factor receptor (EGFR) gene and PTEN; less common secondary malignant gliomas evolve from low-grade tumors (primarily infiltrating astrocytomas, WHO grade II) and typically have TP53 mutations (2,138). Overexpression of p53 and mutations in TP53 have been noted in pediatric malignant glial tumors; EGFR is less commonly expressed (2,164,165).
Supratentorial malignant gliomas arise primarily as cerebral hemispheric tumors; 20-30% present centrally in the thalamus or basal ganglia (144,166). Imaging characteristics are similar to those in adults, with often poorly marginated, peripherally enhancing lesions on MRI or CT associated with surrounding white matter changes (edema); the enhancing components correlate with the cellular, vascularized periphery of the tumor complex (167). Adult studies have shown infiltration of the small, round anaplastic cells well into the perilesional low-density areas on CT or areas of abnormal signal on T2 MRI (2,159,168). The infiltrative characteristics of high-grade gliomas necessitate some caution in aggressive surgery and high-dose local radiation techniques; interest in functional imaging (e.g., PET) for both stereotactic surgical planning and radiation therapy reflects the acknowledged heterogeneity of the tumors and invasiveness beyond areas identified by anatomic imaging (169,170). Even with acknowledged infiltration at a distance from the “overt” tumor, clinical data shows both a direct relationship between the degree of resection and duration of tumor control as well as a pattern of failure that is overwhelmingly at the primary target volume even after high-dose focal radiation therapy (144,160,166,168, 171,172). Leptomeningeal dissemination had been reported in up to 15% of children at diagnosis; however, the large prospective CCG trial (CCG-945) showed disease beyond the primary site only anecdotally (160,168,173).
The diagnosis of high-grade glioma in children has often been challenging to the neuropathologist. Central review of pathology in the CCG-945 trial showed 36% of cases entered based on an institutional diagnosis of anaplastic astrocytomas or glioblastoma were felt to have a discordant diagnosis, primarily a low-grade glioma, based on the reviewers’ interpretation (160,174). As might be anticipated, favorable results of combined modality studies are significantly less impressive when corrected for reviewed histopathology (160,174,175). In addition to confusion with pleomorphic xanthoastrocytoma and some of the benign embryonal tumors in younger children, the newly identified “glioneuronal tumor” may present with an apparently malignant phenotype despite sometimes benign natural history (176).
Only gold members can continue reading. Log In or Register to continue