CT/MRI: proportionate decrease in size of orbit on all imaging modalities. Absent ocular tissue in anopthalmia. Small complete globe in simple microphthalmia; small abnormal globe with coloboma in complex micropthalmia. Globe size abnormalities may be associated with intracranial abnormalities.
Micropthalmia may be associated with coloboma, congenital infections, retinopathy of prematurity, or persistent hyperplastic primary vitreous (PHPV). May see hypertelorism or hyperplastic ethmoid air cells.
Postenucleation/radiotherapy
CT: orbital growth ceases. May get thickening of the orbital walls with time.
Get a greater degree of deformity in younger children.
Fibrous dysplasia
Radiograph/CT: sclerotic ground-glass appearance with thickening and resultant decreased orbital size.
When also involving adjacent facial bones, there is leonine appearance (leontiasis ossea).
Osteopetrosis/thalassemia/severe anaemia
Radiograph/CT: due to hyperostosis.
Mass effect from expansile process in adjacent sinus
CT/MRI: orbital walls are displaced causing deformity and decreased size.
Usually mucocoele or tumor.
Fig. 4.114 Mass effect from expansile process in adjacent sinus. T1-weighted postcontrast coronal MRI shows right frontal ossifying fibroma displacing orbital roof.
Table 4.25 Enlarged orbit
Diagnosis
Findings
Comments
Macrocephaly/hydrocephalus
All imaging modalities: spurious enlargement due to large skull.
CT: thin-slice axial and coronal shows defect in frontal bone. MRI: assessment of cranial contents and associated brain abnormalities.
Interorbital distance is greater than normal.
Mild hypertelorism occurs in multiple syndromes (cleidocranial dysostosis/trisomy 13/Hurler syndrome/thalassemia).
It may be isolated, familial, and of no clinical significance.
Fig. 4.117 Coronal 3D CT shows bicoronal synostosis with harlequin eye appearance and hypertelorism.Fig. 4.118 Coronal CT (bone window) shows nasoethmoidal cephalocele, a defect in the left frontal bone with herniation of intracranial contents into left ethmoid air cells and resultant hypertelorism.Fig. 4.119 T2 axial MRI shows hypertelorism due to nasofrontal cephalocele.
Radiographs/CT: characteristic skull shape with premature fusion of metopic and sagittal sutures.
May see hypoplastic ethmoid sinuses.
Holoprosencephaly
CT/MRI: Midline malformation. Alobar is most severe.
Associated with midface hypoplasia.
Fig. 4.120 AP skull radiograph shows metopic synostosis with me-dial and upward angulation of the orbits and hypotelorism.
Table 4.28 Microphthalmia
Diagnosis
Findings
Comments
Congenital
CT/MRI: Small globe (see Table 4.24). MRI is best imaging modality to assess internal structure of globe, ocular contents, and brain.
Many chromosomal abnormalities: sporadic, AD, recessive, and X-linked forms. One-third of cases have associated cranial malformations. Consider genetic counseling.
Congenital infections (rubella, CMV, syphilis)
CT: Small globe that may contain calcification. Usually have other manifestations of disease; therefore, CT brain indicated to assess for calcification.
CT: Enlarged, elongated globe. Look for sphenoid wing dysplasia and plexiform neurofibroma in NF and characteristic intracranial appearances in SturgeWeber syndrome.
NF: it was thought that plexiform neurofibroma infiltrates angle of ciliary body. Occurs in 50%.
Sturge-Weber syndrome: angiomatous change in ciliary body or angle of anterior chamber.
CT: Non-contrast–enhanced CT (NECT) best for identifying calcification. CECT sees variable enhancement of noncalcified portion of mass and normal sized/enlarged globe.
MRI: Best for extraocular and intracranial disease. Mass hyperintense on T1 and hypointense on T2 relative to vitreous. Postcontrast heterogeneous enhancement. Image whole brain to assess for pineal (trilateral) and suprasellar (quadrilateral) disease.
Calcification rarely seen in other lesions in children < 3 y. Unilateral in 75%. Bilateral and multifocal in 25% is more often familial and must be screened for pineal and suprasellar involvement.
NECT: Microphthalmia, small irregular lens, no calcification, and hyperdense vitreous chamber. Abnormal vitreal tissue may enhance. Linear hyperdensity extending from retina to lens represents Cloquet canal and persistent hyaloid artery.
MRI: High-signal vitreous on T1 and T2, fluid levels as a result of hemorrhage. Also demonstrates canal as previously.
Presents in infancy with leukocoria, microphthalmia, and cataract. Sporadic disease unilateral; bilateral usually part of multisystem syndrome.
CT: High-density vitreous, no calcification, and is indistinguishable from noncalcified retinoblastoma. Vitreous contains wing-shaped retinal detachment. MRI: superior for subretinal effusion—high signal on T1 and T2.
Usually boys, unilateral. Present from birth but present near end of first decade when retina detaches and vision deteriorates.
Retinopathy of prematurity
CT: Noncalcified, retrolental, hyperdense mass that may be associated with retinal detachment. May be calcified and microphthalmic if chronic.
MRI: findings nonspecific.
Uni- or bilateral in setting of appropriate medical history.
Toxocara canis infection
CT: opaque vitreous or a localized irregular, nonenhancing retinal mass. No calcification.
Present at approximate 6 y. History of close contact with dogs and positive serology are suggestive.
Retinal astrocytoma
CT/MRI: intraocular mass (es) with or without exudative retinal detachment, hemorrhage, and calcification.
Rare; isolated or in association with tuberous sclerosis or less commonly NF or retinitis pigmentosa. May be bilateral.
Retinal hemangioma
CT/MRI: Hemorrhage and often retinal detachment.
Post-CE of retina.
Rare.
Medulloepithelioma
CT/MRI: markedly enhancing tumor that may erode the orbital wall or induce hyperostosis.
Rare tumor located anterior to the iris. Occasionally may arise next to the optic nerve. Lack of calcification helps differentiate it from retinoblastoma.
Retinal dysplasia/Norrie disease
CT: microphthalmia and dense vitreous with blood-fluid level. Often also have calcification and retinal detachment.
X-linked recessive, have seizures, mental deficiency and deafness by age 4 y.
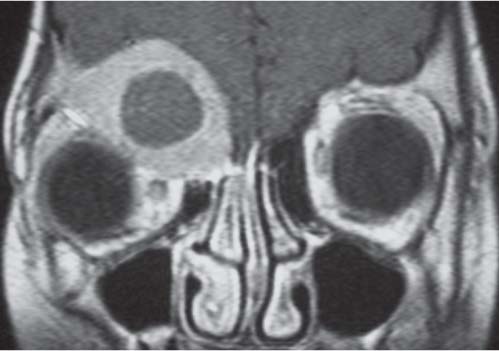
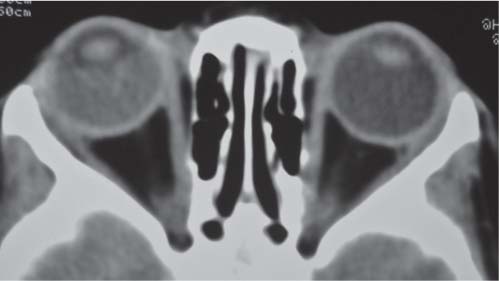
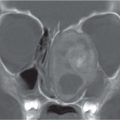
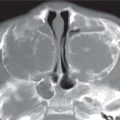
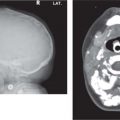
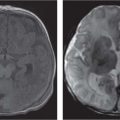
Fig. 4.122 Coronal CT (bone window) shows bilateral calcified intraocular masses consistent with retinoblastomas.Fig. 4.123 T2-weighted fat-saturated sagittal oblique MRI shows intraocular mass with extension into proximal optic nerve and showing T2-weighted hypointensity in keeping with calcification—traits that are characteristic of a retinoblastoma.Fig. 4.124 Persistent hyperplastic primary vitreous. Axial noncontrasted CT shows high-density fluid level in PHPV due to hemorrhage within the right eye.Fig. 4.125 Axial noncontrast CT shows high-density vitreous without focal mass or calcification in Coats disease of the right eye.
Table 4.31 Orbital sclerosis
Diagnosis
Findings
Comments
Fibrous dysplasia
Radiograph/CT: sclerosis, hyperostosis, and bone expansion of one or both orbits. Characteristic ground-glass appearance.
May also have exophthalmos.
LCH
Radiograph/CT: in the healing phase previously lucent lesion will show sclerosis.
Skeletal survey may show other characteristic bone lesions.
Chronic osteomyelitis
Radiograph/CT: sclerosis adjacent to chronically infected frontal sinus.
Osteoma
Radiograph/CT: Well circumscribed sclerotic lesion in frontal sinus. Orbital roof can be displaced and sclerotic.
Osteopetrosis
Radiograph/CT: sclerosing dysplasia with widespread bone involvement.
Skeletal survey will show characteristic appearance of long bones and spine.
Infantile cortical hyperostosis (Caffey disease)
Radiograph/CT: thickening and sclerosis of predominantly upper and lateral orbital walls.
Skeletal survey will show other areas of involvement especially mandible (see Table 4.45).