CHAPTER 52 Otosclerosis
Epidemiology
Genetic, viral, and immunologic factors have been implicated in the etiology of otosclerosis. Studies of familial otosclerosis indicate autosomal dominant transmission with incomplete penetrance. Approximately 60% of patients with otosclerosis have a family history of the disease. Some research points to the measles virus as a possible etiologic agent, with clinical disease developing in those with a persistent measles virus infection and a hereditary predisposition. Other studies suggest that autoimmunity may play a role in the development of otosclerosis.
The disease is most common in the white population and rare in the black population. There is a female preponderance, with a female-to-male ratio of 2:1. It has been reported to be more rapidly progressive in females, and symptoms may worsen during pregnancy.
Clinical Features
Progressive hearing loss, conductive, mixed, or sensorineural, over the course of many years is the primary clinical symptom. Eighty percent of cases are bilateral with onset of hearing loss occurring at ages 15to 45 years. With fenestral otosclerosis, hearing loss is generally conductive, with the otosclerotic plaque impinging of the anterior crus of the stapes, resulting in mechanical stiffness and ultimately fixation. Cochlear otosclerosis usually occurs in conjunction with fenestral disease, and the hearing loss is usually mixed. Rarely, patients may have isolated cochlear otosclerosis and a purely sensorineural hearing loss. Up to 75% of patients may have tinnitus and 25% vestibular symptoms. Otoscopic examination typically reveals a normal tympanic membrane, although 10% of patients may have a reddish hue behind the tympanic membrane, termed Schwartze sign. This sign is related to increased vascularity of the otic capsule in active cochlear otosclerosis.
Pathology
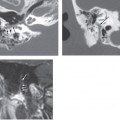
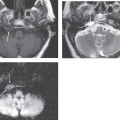
Embryologically, the otic capsule develops from mesenchymal tissue with a cartilaginous framework present at approximately 8 weeks of gestation and enchondral ossification beginning at approximately 16 weeks. Calcification of the otic capsule is complete by 1 year of age. After this, there is normally little bone remodeling and no osteoclastic or osteoblastic activity in the otic capsule. In the early stage of otosclerosis there is pathologic resorption of the enchondral bone of the otic capsule by osteoclasts and vascular proliferation, a process referred to as otospongiosis. In the late stage of disease there is deposition of new bone by osteoblasts, resulting in formation of dense sclerotic bone. Usually the two stages coexist. The result is areas of disorganized bone that do not follow the normal contours of the otic capsule. This abnormal bone can extend into the middle ear or the perilymphatic spaces.
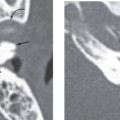
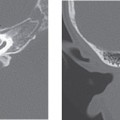
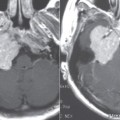
Fenestral otosclerosis involves the bone just anterior to the oval window, a location corresponding to the embryologic fissula ante fenestram. The otosclerotic plaque impinges on the anterior crus of the stapes, resulting in mechanical stiffness and conductive hearing loss. With progression of disease, complete fixation of the stapediovestibular articulation or replacement of the oval window by otosclerotic bone may occur. Progressive disease may extend posteriorly along the oval window to involve the fossula post fenestram. Cochlear (retrofenestral) otosclerosis involves the bone surrounding the cochlea. It is hypothesized that diffusion of cytotoxic enzymes from the otosclerotic foci into the cochlear fluid produces the sensorineural hearing loss. Cochlear otosclerosis is rarely isolated and is usually associated with fenestral otosclerosis.
Treatment
Treatment of the conductive hearing loss is primarily surgical. The exact surgical procedure is based on the extent of disease and may vary from stapes mobilization, partial stapedectomy, stapedotomy, to complete stapedectomy and prosthesis placement.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


