and Anca Dragomir2
(1)
School of Health and Medical Sciences, Örebro University, Örebro, Sweden
(2)
Department of Pathology, Uppsala Academic Hospital, Uppsala, Sweden
Abstract
X-ray microanalysis conducted using the scanning electron microscope is a technique that allows the determination of chemical elements in bulk or semi-thick specimens. The lowest concentration of an element that can be detected is in the order of a few mmol/kg or a few hundred parts per million, and the smallest amount is in the order of 10−18 g. The spatial resolution of the analysis depends on the thickness of the specimen. For biological specimen analysis, care must be taken to prevent displacement/loss of the element of interest (usually ions). Protocols are presented for the processing of frozen-hydrated and freeze-dried specimens, as well as for the analysis of small volumes of fluid, cell cultures, and other specimens. Aspects of qualitative and quantitative analysis are covered, including limitations of the technique.
Key words
X-ray microanalysisScanning electron microscopyPeak-to-background ratioFrozen-hydrated specimenSephadex beadsCell cultureAirway surface liquid1 Introduction
X-ray microanalysis is a technique of elemental analysis that is conducted in an electron microscope. In this chapter, only X-ray microanalysis conducted in a scanning electron microscope will be discussed. The technique is based on the generation of characteristic X-rays in atoms of the specimen by the incident beam electrons (see Fig. 1). When an incident electron hits atoms in the specimen, this may cause an inner shell ionization, that is, the incident electron transfers so much energy to an electron in one of the inner shells of the atom that this electron leaves its shell and the atom. This results in a “gap” in the shell where this electron originally was present. This situation is unstable and may be remedied by an electron transition, in which an electron from a higher shell falls back to fill the gap. Because this electron has a higher energy than the electron originally removed, energy is liberated in this electron transition in the form of an X-ray. This X-ray has an energy that is equal to the energy difference between the shells between which the electron transition takes place. These X-rays are characteristic for the element from which they originate and hence contain information on which elements are present in the specimen (and how much of each).
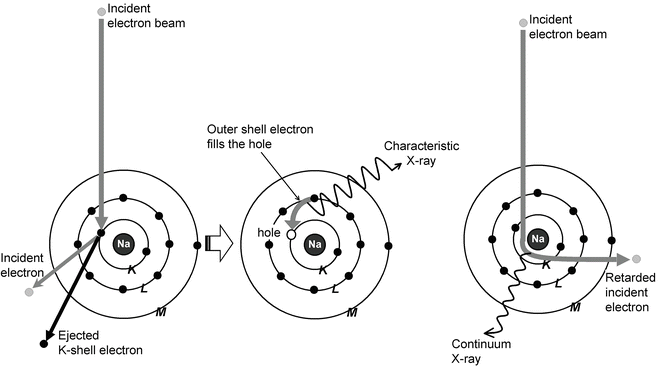
Fig. 1
Generation of characteristic and continuum X-rays in atoms of the specimen by incident electrons
Because there are many electron shells and subshells, each with their particular energy, there are, in principle, many potential possible electron transitions. If the gap occurs in the K-shell, the resulting X-ray is called a K-line; if the gap occurs in the L-shell, the resulting X-ray is called an L-line, and so on. If the gap in the K-shell is filled by an electron coming from the L-shell, the resulting X-ray is called a Kα-line; if it is filled by an electron coming from the M-line, the resulting X-ray is called a Kβ-line. Not all electron transitions are equally probable; hence the Kα-line is about five times as intense as the Kβ-line; some theoretically possible electron transitions do not occur at all. The Kβ-line has a slightly higher energy than the Kα-line, since the M-shell has a higher energy than the L-shell, and so the energy difference between M- and K-shells (E M − E K) is more than E L − E K. Therefore, the energy of the Kβ-line of a particular element is higher than that of the Kα-line of the same element. The energy of an L-line from a particular element is always lower than that of the K-lines of that element, but higher than that of the M-lines from the same element, since E M − E L is less than E L − E K, and so on.
In addition to characteristic X-rays, the beam electrons generate background or continuum X-rays, which are generated by the incident electron being retarded by the electric field of the nucleus of the atom. The energy loss that is suffered by the incident electron in this process is released in the form of a background or white X-ray (see Fig. 1). This white or background radiation does not contain any information about the chemical identity of the element from which this radiation originates, but the intensity of the radiation is related to the total mass of the analyzed volume in the specimen and is used in quantitative analysis [1, 2]. A typical X-ray spectrum, showing characteristic peaks and background, is shown in Fig. 2. The lowest concentration of an element that can be detected is in the order of 10−18 g. The spatial resolution of the analysis depends on the thickness of the specimen. The best spatial resolution is obtained in (thin) sections where as a rule of thumb the diameter of the analyzed volume is about half the section thickness. If bulk specimens are analyzed, as is done in the scanning electron microscope, the spatial resolution depends on the accelerating voltage and the composition of the specimen. A typical value for analysis of freeze-dried biological material at an accelerating voltage of 20 kV is approx. 10 μm [1, 3].
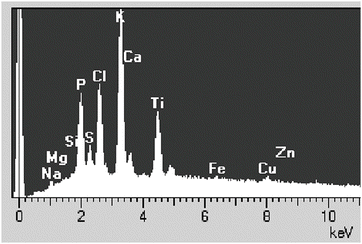
Fig. 2
Example of an X-ray spectrum with characteristic peaks and background
The scanning electron microscope produces a narrow beam by demagnification in the condenser/final lens system of the beam leaving the filament. This beam can be directed to the feature of interest. However, as soon as the incident electrons hit the specimen surface, they collide with atoms in the specimen and are spread under a certain angle. Hence, an interaction volume is created from which X-rays may be generated. The size of this interaction volume depends on the density and composition of the specimen and on the energy of the incident electron (determined by the accelerating voltage) (see Fig. 3). X-rays may be generated throughout the interaction volume.
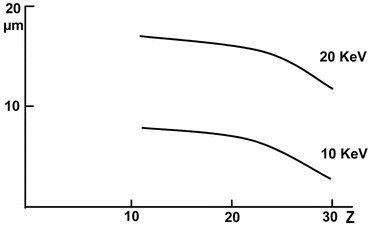
Fig. 3
Dependence of the interaction volume on the atomic number and the accelerating voltage
The scanning electron microscope also allows the specimen to be visualized. As will be discussed below, specimens for X-ray microanalysis are generally not coated with gold or impregnated with heavy metals, so the morphological information from the specimen is not optimal. However, at the level of resolution required, it is generally sufficient to coat the specimen with a conductive carbon layer, to avoid charging.
2 Materials
1.
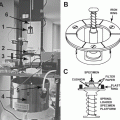
Scanning electron microscope is equipped with an X-ray detection system and, optionally, with a cryo-chamber with cold stage and low-temperature carbon-coating devices (see Fig. 4 and Note 1).
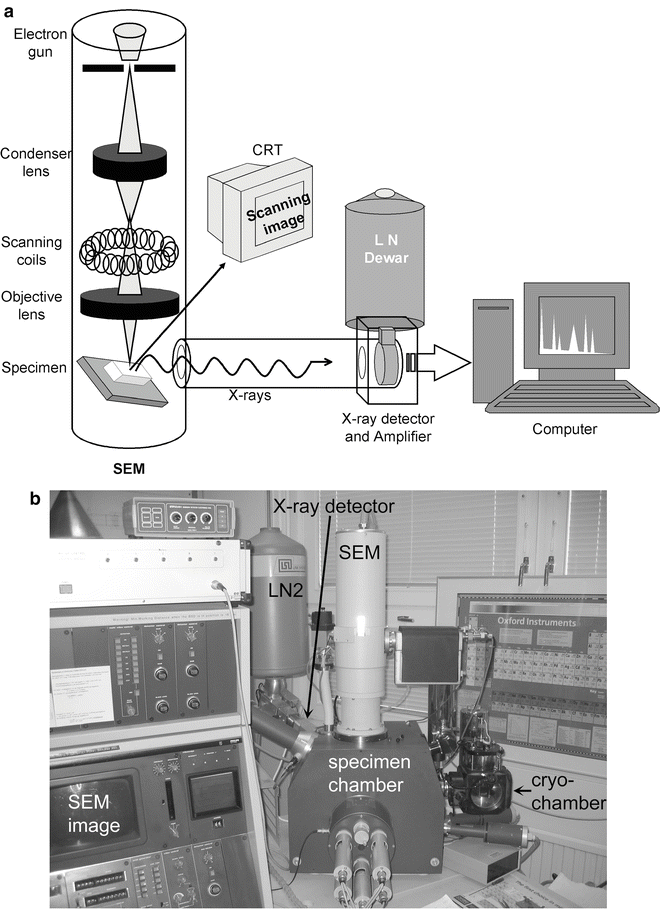
Fig. 4
(a) Schematic of the electron microscope with the X-ray detection system. (b) Photograph of the specimen chamber with X-ray detector
2.
Freezing device, liquid nitrogen, and pressurized propane or ethane for rapid freezing of the specimen (see Note 2).
4.
Planchets of spectroscopically pure carbon for scanning electron microscopy.
5.
Sephadex G-25 beads, diameter 20–40 μm (Sigma-Aldrich, St. Louis, MO).
6.
Hydrophobic volatile silicon oil (Dow Corning 200/lcS, BDH, Poole, UK).
7.
Nylon electron microscopy grids.
8.
Sterile cellulose nitrate or cellulose nitrate/cellulose acetate filters, or another suitable substrate for cell culture.
10.
Double-sided tape.
11.
Filter paper.
12.
Salt solutions, for example, NaCl or KCl, ranging from 50 to 250 mM.
13.
Rinsing solutions, e.g., 150 mM ammonium acetate or 300 mM mannitol in distilled water.
3 Methods
3.1 General Considerations
Most scanning electron microscopes operate at vacuum conditions; therefore, specimen preparation has to take this into account. In a conventional scanning electron microscope, specimens therefore have to be dried; alternatively, frozen-hydrated specimens can be used. It is, however, possible to conduct X-ray microanalysis in an environmental scanning electron microscope at atmospheric pressure [4].
It should be realized that glutaraldehyde fixation will cause leakage of diffusible ions, and it has been shown that nearly all Na, K, Cl, Mg, and Ca are lost during such treatment. Hence, unless the interest is restricted to very firmly bound elements, cryotechniques have to be used in the preparation of specimens for X-ray microanalysis:
1.
The first step of a cryotechnique is the rapid freezing of the specimen. Because analysis in the scanning electron microscope is not carried out at high resolution, requirements for freezing are somewhat less stringent than when X-ray microanalysis is carried out at high resolution in a (scanning) transmission electron microscope. Similar freezing techniques can be used: freezing using a liquid coolant or freezing against cold metal. As liquid coolants, liquid propane or ethane cooled by liquid nitrogen, or liquid nitrogen itself, can be used. Of these, liquid propane or ethane is to be preferred (see Note 2).
2.
After the specimen has been frozen, several avenues can be chosen. It is possible to fracture the specimen and to analyze it in the frozen-hydrated state [5, 6]; alternatively, after fracturing, the specimen can be dried and analyzed in the freeze-dried state [7]. It is also possible to prepare thick cryosections of the specimen in a conventional cryostat; these cryosections can then be dried and analyzed [3, 8]. A type of specimen that is suitable for analysis in the scanning electron microscope is cultured cells that can be grown on a solid substrate, frozen and freeze-dried, and then analyzed [9]. Small amounts of fluid can be collected in situ by placing Sephadex beads into the fluid covering, for example, an epithelial surface [10, 11]; these beads with the collected fluids are then dried and analyzed. This does not exhaust the types of specimen that can be analyzed in the scanning electron microscope: any type of specimen that can be viewed in a scanning electron microscope can be the subject of X-ray microanalysis, if the general rules above are followed.
3.2 Cryofractured Frozen-Hydrated and Frozen-Dried Specimens
Analysis of frozen-hydrated specimens has two advantages over analysis of frozen-dried specimens: (1) it allows analysis of large fluid-filled compartments and spaces, for instance, the lumina of glands and ducts, or fluid-filled spaces in the inner ear, and (2) it allows the elemental concentrations to be related to the wet weight of the specimen, which is physiologically more informative than the relation to the dry weight; by carrying out the analysis first in the frozen-hydrated state and then in the freeze-dried state, the ionic concentrations can be related to the water in the analyzed compartment, which physiologically speaking is the ultimate relevant parameter.
The procedure consists of the following steps:
1.
Rapid freezing.
2.
Transfer to the cold stage of the scanning electron microscope.
3.
Fracturing to expose an internal surface.
4.
Analysis at low temperature, typically close to −190 °C.
5.
Step 4 may be followed by freeze-drying inside or outside the scanning electron microscope.
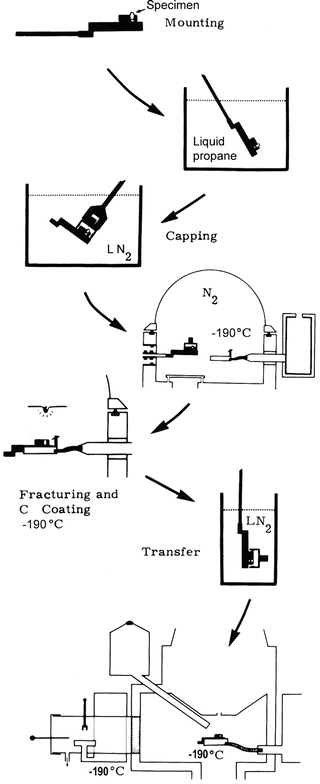
As discussed previously, freezing should be conducted as rapidly as possible. Marshall and colleagues [5, 6] suggest freezing in melting nitrogen, but other methods, such as freezing in liquid propane or ethane, or against metal cooled by liquid nitrogen, should be adequate alternatives. After the freezing step, the specimen should be transferred to liquid nitrogen if it is not already in this medium. Fracturing at low temperatures (−170 °C or lower) provides, according to Marshall and colleagues [5, 6], a rough-surfaced specimen with sufficient topographic detail. At higher temperatures, a smoother fracture surface is obtained. Although this surface may have some advantages over a rough surface (a smooth surface will show less variation in X-ray intensity with topography), smooth surface provide less topographic information, and it is more difficult to localize features of interest on them. Frozen-hydrated specimens charge in the electron beam, but this problem can be reduced by coating the specimen with a conductive carbon layer. Most commercially available cold stages allow one to conduct coating with carbon at low temperature. Marshall and colleagues [5, 6] state that the analysis of these specimens should be performed with a rastered beam (preferably covering a size of 25 μm2), rather than with a stationary beam (see Note 4). An example of a frozen-hydrated specimen of rat trachea in cross section is illustrated in Fig. 6.

Fig. 6
Scanning electron micrograph of a thick cryosection of pig trachea. The layer of airway surface liquid (ASL) is easily identifiable [15]
If one wishes to continue the analysis in the frozen-dried state, the specimen can be freeze-dried on the cold stage without breaking the vacuum, by raising the temperature. The specimen is then analyzed again in the freeze-dried state, and the water content of the specimens (the water fraction F w) can be calculated from
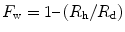
where R h and R d are the relative intensities (peak-to-background ratios) in the hydrated and dried state, respectively, and, once the water fraction F w is known, the elemental concentrations related to the cell water can be calculated from there [2].
(1)
3.3 Thick Freeze-Dried Cryosections
To simplify the quantitative procedure, the cryosections should be infinitely thick to the electron beam, i.e., the beam should not penetrate the section completely. Because a 20-kV beam penetrates freeze-dried biological soft tissue to approx. 10–12 μm, sections of tissue to be analyzed at 20 kV should be at least approx. 16 μm thick. Different values may apply if the analysis is conducted at a different accelerating voltage or if hard (calcified) tissue with a greater density is to be analyzed. The procedure is carried out as follows [3, 8, 12]:
1.
The tissue is rapidly frozen and mounted on the specimen holder of a cryostat. The cryostat is set at a temperature of −20 to −30 °C.
2.
Cryosections (approx. 16 μm thick of soft tissue for analysis at 20 kV) are cut and mounted on a planchet of spectroscopically pure carbon (see Note 5).
3.
Leave the carbon planchet for 48 h in the cryostat with a copper weight on top of the section. Then, remove the copper weight and store the planchet with the section in a desiccator over silica gel or another drying agent until analysis.
4.
Coat the sample with carbon before analysis. Mount the carbon planchet onto the specimen stage of the scanning electron microscope and analyze at room temperature at 20 kV for 100–200 s.
An example of a scanning electron micrograph of a cryosection of freeze-dried biological tissue (human muscle) is shown in Fig. 7.
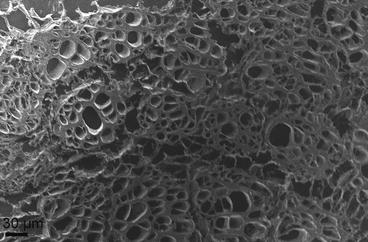
Fig. 7
Scanning electron micrograph of a thick cryosection of human muscle. The “holes” in the cells are due to ice crystal formation during freezing
3.4 Small Volumes of Fluid Collected on Sephadex Beads
A method that can be used to collect fluid from the surface of epithelia (e.g., uterine epithelium or airway epithelium in experimental animals or in humans) uses dextran (Sephadex G-25) beads (diameter 20–40 μm; see Fig. 8). The following procedure is used [11, 13–15]:

Fig. 8
Scanning electron micrograph of a Sephadex G-25 bead used to collect fluid
1.
The beads are equilibrated for 20 min with the fluid, for example, in the uterus or trachea by spreading the beads over the epithelial surface.
2.
Then, the beads are collected and washed with hydrophobic volatile silicone oil to remove adhering fluid and debris.
3.
Each bead is individually moved to a nylon electron microscopy grid, which had been submerged into the oil, until the grid contains at least 15 beads.
4.
Alternatively, the Sephadex G-25 beads can be applied to double-sided tape attached to small pieces of filter paper (approx. 1 × 1 mm2).
5.
The filter papers with the beads are placed onto the epithelium (beads facing downwards) for 20 min. Saturation of the beads with a salt solution is obtained after 5 min [13].
6.
 Microwave-Assisted Processing and Embedding for Transmission Electron Microscopy
Microwave-Assisted Processing and Embedding for Transmission Electron Microscopy
 Electron Microscopy of Microtubule Cytoskeleton Assembly In Vitro
Electron Microscopy of Microtubule Cytoskeleton Assembly In Vitro
 Biological Applications of Energy-Filtered TEM
Biological Applications of Energy-Filtered TEM
 Freeze Fracture and Freeze Etching
Freeze Fracture and Freeze Etching
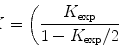 Elemental and Isotopic Imaging of Biological Samples Using NanoSIMS
Elemental and Isotopic Imaging of Biological Samples Using NanoSIMS
 FIB-SEM Tomography in Biology
FIB-SEM Tomography in Biology




Then, the filter paper with saturated beads is removed and washed with hydrophobic volatile silicone oil to remove adhering fluid and debris. Each bead is individually moved to a nylon electron microscopy grid as described above. The grid with beads is slowly lifted out of the oil bath and mounted onto an aluminum holder covered with carbon adhesive tape and left at room temperature for evaporation of the oil.
Related posts:
Microwave-Assisted Processing and Embedding for Transmission Electron Microscopy
Electron Microscopy of Microtubule Cytoskeleton Assembly In Vitro
Biological Applications of Energy-Filtered TEM
Freeze Fracture and Freeze Etching
Elemental and Isotopic Imaging of Biological Samples Using NanoSIMS
FIB-SEM Tomography in Biology
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
