CHAPTER 39 Phakomatoses: Tumor Suppression Gene Defects
The phakomatoses, or neurocutaneous syndromes, include neurofibromatosis types 1 and 2 (NF1 and NF2), tuberous sclerosis complex (TSC), von Hippel-Lindau disease (VHL), ataxia-telangiectasia, and basal cell nevus syndrome (BCNS). All these disorders show a high frequency of tumor development. NF1, NF2, TSC, and VHL are attributed to a variety of tumor suppression genes. Ataxia-telangiectasia belongs to a group of chromosome breakage syndromes that result from deficient DNA repair. BCNS is caused by derangement of the hedgehog signaling pathway. Of surprise is the fact that the signaling pathway with a major role in early embryonic development is also responsible for tumor development.
NEUROFIBROMATOSIS TYPE 1
NF1 is a common genetic disorder characterized by multiple tumors in the central and peripheral nervous systems, cutaneous pigmentation, and lesions of the vascular system and viscera. Additionally, there is a tendency for a variety of tissues to undergo malignant transformation.1 It is also referred to as von Recklinghausen’s disease, peripheral neurofibromatosis, and classic neurofibromatosis.
Epidemiology
NF1 is one of the most common genetic disorders, with an estimated prevalence of 1 in 2000 to 5000 individuals in most population studies, regardless of gender and ethnic background.2
Clinical Presentation
• Six or more café-au-lait spots whose greatest diameter is more than 5 mm in prepubertal patients and more than 15 mm in postpubertal patients
• Two or more neurofibromas of any type, or one or more plexiform neurofibromas
• Freckling of the axillary or inguinal region (Crowe’s sign)
• Two or more Lisch nodules (iris hamartoma)
• A distinctive osseous lesion such as sphenoid wing dysplasia or thinning of the cortex of the long bone (with or without pseudarthrosis)
• A first-degree relative (parent, siblings, or offspring) with NF1 according to the previously mentioned criteria
Only about half of children with NF1 meet the NIH criteria for diagnosis by 1 year of age, but almost all do so by age 8 because many features of NF1 increase in frequency with age.3 Other clinical characteristics of NFI include learning disabilities, short stature, macrocephaly, and predisposition to developing neoplasia, such as gliomas, malignant peripheral nerve sheath tumors (MPNST), and myeloid malignancies.
Neurofibromas are classified as cutaneous (Fig. 39-E1A), subcutaneous, and plexiform, with the first two usually occurring in early adolescence and the latter being congenital (at least the diffuse type). Plexiform neurofibromas can become larger, progress to malignancy (usually MPNST), and may occur almost anywhere in the body. They commonly develop in the orbit and cause impaired ocular movement and exophthalmos. The neck is another common location.
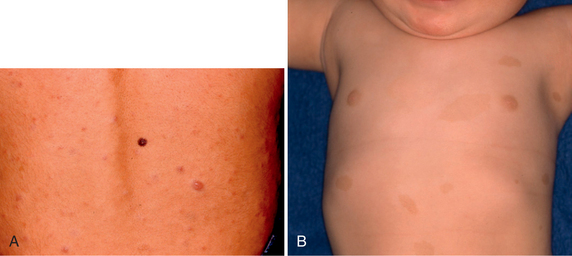
FIGURE 39-E1 Neurofibromatosis type 1. Skin manifestations. A, Cutaneous neurofibromas. B, Café-au-lait spots.
Café-au-lait spots are benign pigmented lesions and are the first manifestation of NF1, usually becoming evident during the first year of life (Fig. 39-E1B). Freckling of the axilla will develop later in about two thirds of patients. Lisch nodules of the iris are best discernable by slit lamp examination. They begin to appear in childhood and are finally present in all NF1 patients.4
Skeletal manifestations include kyphoscoliosis, overgrowth or undergrowth of bones, bony rarefactions due to adjacent neurofibroma, and pseudarthrosis of the tibia.1,5 Skull lesions include macrocephaly, lambdoid suture defect, and dysplasia of the greater sphenoidal wing.
Neurologic manifestations can be grouped into five major categories, including cognitive disabilities, intracranial tumors, tumors of the peripheral nerves, spinal nerve tumors, and cerebral infarction.1 Thirty to 60 percent of children with NF1 have learning disabilities, and they tend to do better on verbal tasks than on performance tasks. Only 8% of patients with NF1 were reported to have an IQ less than 70.1 Intracranial tumors include optic pathway gliomas, brain stem gliomas, and other hemispheric tumors. Tumors of the peripheral nerves are mainly neurofibromas, can arise at any age, and can involve any major nerve. Spinal nerve tumors generally develop slowly and are rare in children. Cerebral infarctions result from cerebrovascular occlusive disease, most commonly affecting the supraclinoid portion of the internal carotid artery or one of its major branches.1
Optic pathway glioma is the most common tumor of the central nervous system (CNS) in patients with NF1. It usually develops in the first 2 decades, with a peak incidence at around age 4 to 5 years. Although the reported incidence ranges from 15% to 23%, only approximately one half of the affected patients develop signs and symptoms.1 The tumor may involve any portion of the optic pathway. Although optic pathway gliomas are an incidental finding in the majority of the children with NF1, they may result in progressive compromise of visual activity and may cause serious symptom. Precocious puberty may occur if the tumor extends to the hypothalamus. Spontaneous regression may occur and may be related to the NF1 gene activity as tumor suppressor. Therefore, aggressive therapy is not indicated without clinical or radiologic evidence of progression.1,4,5 Astrocytomas may develop in the mesencephalic tectum, brain stem (especially medulla), cerebellum, and cerebral hemispheres.5 NF-related brain stem gliomas show clinical courses different from those of isolated brain stem gliomas. They tend to have indolent courses and better long-term outcome. Although appropriate management for both optic pathway gliomas and other gliomas is still under discussion, conservative management with serial follow-up MRI is recommended.4,5
Another CNS manifestation in NF1 is bilateral, nearly symmetrical zones of myelin vacuolization (ZMVs) that appear as areas of hyperintensity on T2-weighted (T2W)/fluid-attenuated inversion recovery (FLAIR) images in 60% to 70% of patients. The details of MRI findings are discussed later in the section on imaging. These zones become apparent after infancy and tend to diminish or disappear with age. They have been given various names, such as histogenetic foci, neurofibromatosis bright objects, and hamartomatous lesions. Because these lesions are pathognomonic, several authors proposed to include them in the diagnostic criteria for pediatric patients. Their relationship with cognitive disabilities has been investigated by several authors and is still controversial. The anatomic location of ZMVs may be more important than their number. Particularly, myelin vacuolization in the thalamus might significantly correlate to neurophysiologic disability.5,6
The value of performing brain MRI for NF1 patients at the time of clinical diagnosis is controversial. It is useful in helping to establish the diagnosis in some and in identifying complications before they become clinically apparent in others, but clinical management is not affected by findings of intracranial lesions such as optic nerve thickening and ZMVs in asymptomatic patients.3
Pathophysiology
NF1 is transmitted in an autosomal dominant inheritance pattern with complete penetrance and variable expressivity. The disease is presumed to result from a loss-of-function mutation of the NF1 gene. Approximately 50% of cases show de novo mutations. The reason for such a high mutational rate in the NF1 gene remains unknown but might be due to its large size, genomic position, and unusual structure. Over 80% of new mutations have been found to be paternal in origin. Although more than 500 different germline NF1 mutations have been reported, there have been few consistent relationships between the clinical phenotype and size, type, and location of the gene.1,7 Currently, only two clear correlations have been observed: (1) a whole NF1 gene deletion presents large numbers and early appearance of cutaneous neurofibromas, more frequent and more severe cognitive abnormalities, and sometimes somatic overgrowth with large hands and feet, and dysmorphic facial features; and (2) a 3-bp in-frame deletion of exon 17 does not present surface plexiform neurofibromas despite typical pigmentary features in NF1.
The NF1 gene is mapped to chromosome 17q11.2 and is a tumor suppressor gene. It encodes neurofibromin, a cytoplasmic protein that is predominantly expressed in neurons, Schwann cells, oligodendrocytes, astrocytes, and leukocytes. The function of neurofibromin is not fully understood, but it appears to activate RAS guanosine triphosphatase (GTPase), thereby controlling cellular proliferation and acting as a tumor suppressor. Neurofibromin probably has other functional properties as well, including regulation of adenylylcyclase activity and intracellular cyclic adenosine monophosphate (AMP) generation. The pathogenic mechanisms of NF1 gene mutations have been widely investigated, particularly focusing on tumor formation. Neurofibromas are considered to occur as a result of loss of the remaining wild-type NF1 allele. Loss of heterozygosity during fetal development may result in the formation of plexiform neurofibromas, whereas a second-hit mutation later in life could lead to development of cutaneous neurofibromas. Molecular studies for MPNSTs from NF1 patients have shown several tumor suppression gene mutations in addition to NF1 mutations, and these findings suggest that a second genetic event is required for the MPNST formation in NF1 patients.7 In NF1 pilocytic astrocytomas (optic pathway tumors), no detectable neurofibromin expression or increased RAS activity is found. These reports suggest that astrocytoma formation in NF1 patients requires a second genetic hit (loss of NF1 expression).7
Pathology
There are few pathologic reports on the histology of the bilateral symmetrical zones of hyperintensity. In these, myelin vacuolization and spongiform myelopathy have been reported.8
Imaging
Optic Pathway Gliomas and Other Gliomas
Astrocytomas are more common in NF1 patients than in the general population.4 Although optic pathway gliomas (Figs. 39-1 and 39-2) are the most common, astrocytomas may occur in the mesencephalic tectum, brain stem (Fig. 39-3), cerebellum, and cerebral hemispheres (see also Fig. 39-E2).5 Imaging manifestations of these gliomas in NF1 patients are identical to those of patients without NF1.
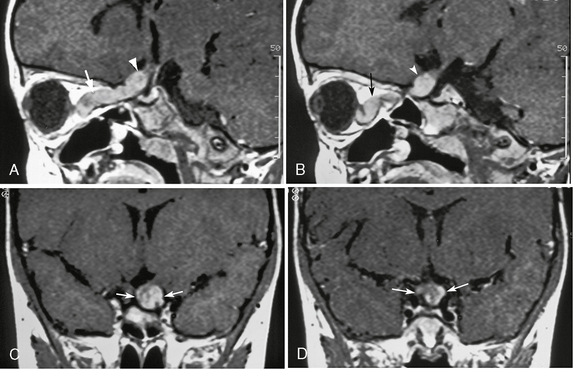
FIGURE 39-1 Neurofibromatosis type 1. Optic pathway glioma in a 3-year-old girl is evident on gadolinium-enhanced T1W sagittal (A, B) and coronal (C, D) MR images. Enlargement of the left optic nerve is seen in the intraorbital portion (arrows, A, B), prechiasmatic portion (arrowheads, A, B), and the optic chiasm (arrows, C, D). The tumor shows diffuse enhancement, and the optic nerve is markedly tortuous.
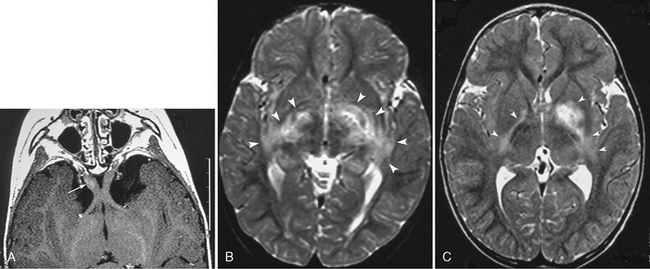
FIGURE 39-2 Neurofibromatosis type 1. Optic pathway glioma in a 2-year-old boy as shown on gadolinium-enhanced T1W axial (A) and T2W axial (B, C) MR images. Prechiasmatic portion of the right optic nerve shows mild enlargement and faint enhancement (arrow, A). T2W images reveal extended hyperintensity in the globi pallidi, internal capsule, and the optic tracts (arrowheads, B, C). It is impossible to determine whether this is a glioma or a zone of myelin vacuolization.
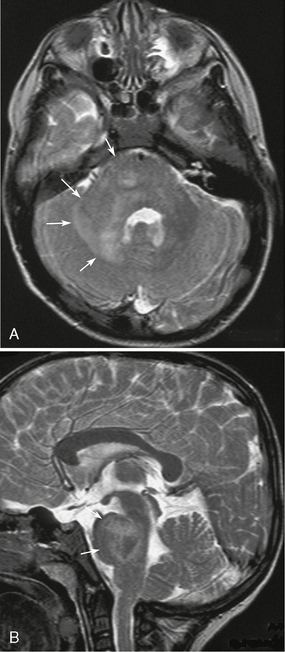
FIGURE 39-3 Neurofibromatosis type 1. Brain stem glioma or unidentified bright objects in a 4-year-old boy as shown on axial (A) and sagittal (B) T2W MR images. Enlargement and ill-defined heterogeneous hyperintensity are seen in the pons, right middle cerebellar peduncle, and cerebellar white matter (arrows, A, B). It cannot be determined if this is a glioma or a zone of myelin vacuolation.
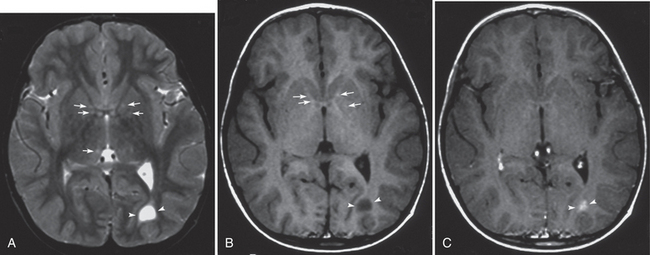
FIGURE 39-E2 Neurofibromatosis type 1. Coexisting tumoral and hamartomatous lesions and zones of myelin vacuolization (ZMVs) in a 4-year-old boy. Axial T2W (A), T1W (B), and gadolinium-enhanced T1W (C) MR images. Several small ZMVs in the globi pallidi and the thalami are seen as slight hyperintense foci on a T2W image (arrows, A). The foci in the globi pallidi show slight T1 shortening (arrows, B). An astrocytoma appears in the left occipital deep white matter as a well-defined markedly T1- and T2-prolonged lesion with gadolinium enhancement (arrowheads, A-C).
MRI
MRI is the best modality for assessing both intracranial and extracranial involvement of the optic pathway. T1-weighted (T1W) images particularly show distortion, enlargement, and sometimes elongation of the optic nerves, optic chiasm, and/or optic tracts (see Fig. 39-1). T2W/FLAIR sequences show abnormal hyperintensity extending posteriorly to the lateral geniculate bodies (see Fig. 39-2). The signal intensity of the lesions is variable on both T1W and T2W images. Enhancement of the lesions also is variable.4,5
Brain stem gliomas may be indistinguishable from UBOs. Both show high intensity on T2W images and may show mass effect without enhancement (see Fig. 39-3). Therefore, follow-up MRI is mandatory. Apparent diffusion coefficient (ADC) could be reduced in the area of high-grade neoplasm.
Magnetic Resonance Spectroscopy
Proton MR spectroscopy (MRS) can help in differentiating between normal brain, ZMVs, and glioma.5 On multivoxel proton MRS, ZMVs have shown elevated choline (Cho) levels, possibly reflecting increased myelin turnover; reduced creatinine (Cr); and normal N-acetyl-aspartate (NAA). Neoplasms show a Cho/Cr ratio greater than 2 and no NAA.9 MRS may show metabolic abnormality even in normal-appearing areas of the brain,5,9 and ADC is significantly higher in normal-appearing areas than in control subjects.4,10
Zones of Myelin Vacuolization
ZMVs are the most common intracranial lesions, occurring in about two thirds of NF1 patients and approximately 75% of children with NF1. The incidence rises to 90% in patients with optic pathway glioma (see Fig. 39-2). Because a pathologic analysis has shown that they reflect myelin vacuolization, it is speculated that dysplasias of myelin are the cause of these lesions.4 They are characteristic findings and appear to be pathognomonic, especially in pediatric patients.
ZMVs are typically multiple and commonly located in the white matter, splenium of the corpus callosum, hippocampi, basal ganglia (especially in the globus pallidus), internal capsules, thalami, brain stem, middle cerebellar peduncles, and cerebellar hemispheres (see Figs. 39-2 and 39-4).
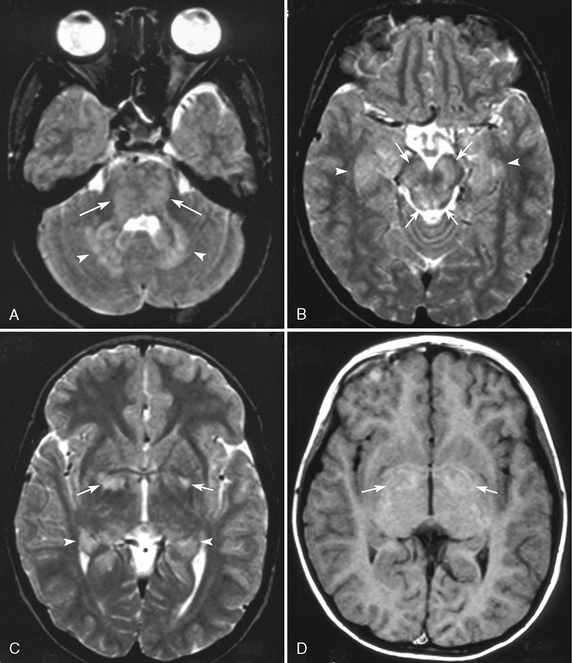
FIGURE 39-4 Unidentified bright objects in a patient with neurofibromatosis type 1 evident on axial T2W (A-C) and T1W (D) MR images. T2W images show multiple hyperintense lesions in the pons (arrows, A), cerebellar dentate nuclei (arrowheads, A), and the midbrain (arrows, B). Bilateral hippocampi are slightly thickened and show T2 prolongation (arrowheads, B, C). Bilateral globi pallidi show as signal hyperintensity on a T2W image (arrows, C), and the peripheral areas of T2 prolongation are slightly hyperintense on a T1W image (arrows, D).
CT
CT is not effective for detecting ZMVs, although a few of them can present as low-density lesions.
MRI
ZMVs are hyperintense on T2W, proton-density–weighted, and FLAIR images. They are usually isointense to slightly hypointense on T1W images (see Figs. 39-2 to 39-4) but sometimes may be hyperintense, particularly in the globus pallidus (see Fig. 39-4D and 39-E2B). Slight T1 shortening can also be observed in lesions of the brain stem and thalami. This has been suggested to relate to the presence of ectopic Schwann cells and melanocytes, to hypermyelination in hamartomatous or gliotic areas, to repair of vacuolized regions11 and to microcalcifications.8 But it tends to develop after the T2 prolongation appears and before the T2 prolongation disappears. This may mean that T1 shortening reflects delayed reactive formation of myelin.11
In general, ZMVs show neither contrast enhancement nor surrounding edema; yet mass effect may be present and transient enhancement has been reported in a few cases. Therefore, the differential diagnosis between myelin vacuolization and a low-grade neoplasm is difficult (see Figs. 39-2 and 39-3), so follow-up MRI is necessary. A neoplasm should be suspected when the lesion shows clear low signal with contrast enhancement on T1W images (see Fig. 39-E2).5 Moreover, the biologic behavior of ZMVs cannot be predicted by imaging alone.4 Benign-appearing lesions can evolve into neoplasms, whereas neoplastic-like lesions can decrease in size or disappear.
ZMVs are typically not found in infancy, increase in number and size until adolescence, and then attenuate, which might reflect myelin repair.4,11
Apparent diffusion coefficients (ADCs) are reported to be increased in ZMVs as compared with other areas of the brain in NF1 patients. Even the normal-appearing brain shows higher ADC values in patients with NF1 than in control individuals.4,10
Other Brain Lesions
MRI
Many NF1 patients have an increased volume of cerebral white matter with a large corpus callosum (Fig. 39-E3). These findings are postulated to result from increased size and number of axons.
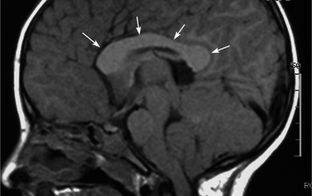
FIGURE 39-E3 Callosal thickening of neurofibromatosis type 1. A midsagittal T1W MR image shows diffuse thickening of the corpus callosum (arrows).
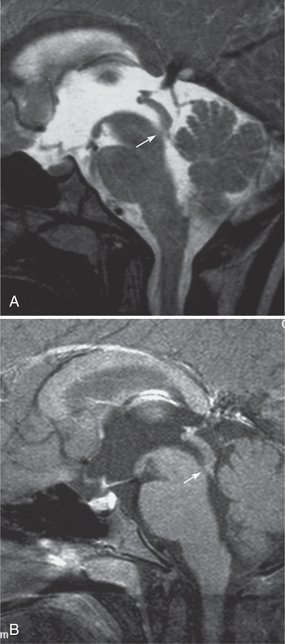
Hydrocephalus is occasionally seen in NF1 patients. The causes of hydrocephalus include brain stem neoplasms adjacent to the aqueduct and benign aqueduct stenosis (Fig. 39-E4).4
Orbital and Calvarial Manifestations/Plexiform Neurofibromas
CT
Bone dysplasias of the head and neck are common in NF1 patients and include defects of the sphenoid wing (Fig. 39-5A) and along the lambdoid suture (Fig. 39-E5). The latter has no clinical significance. Unilateral dysplasia of the sphenoid wings is characterized by hypoplasia or partial agenesis of the greater wing and partly of the lesser wing. As a result, the middle cranial fossa expands and the temporal lobe bulges through the bone defect. Pulsation of the temporal lobe may cause progressive pulsatile exophthalmos.4,5 Rarely, there is enophthalmos instead.
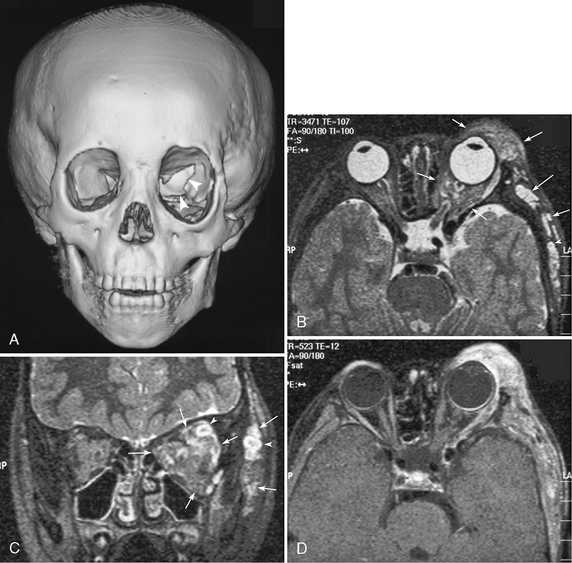
FIGURE 39-5 Neurofibromatosis type 1. Dysplasia of the left sphenoid wing with plexiform neurofibromatosis shown on a 3D CT image of the face and skull (A) and on T2W axial (B), coronal (C), gadolinium-enhanced T1W (D) MR images. Partial agenesis of the left greater sphenoid wing (arrowheads, A) and secondary expansion of the left orbit can be seen on CT. T2W images show multiple nodular and diffuse hyperintense lesions in the left orbit and extracranial region (arrows, B, C). Some nodular lesions show the target appearance (arrowheads, B, C). The lesions also show heterogeneous enhancement (D).
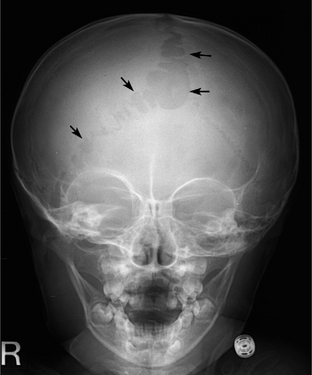
FIGURE 39-E5 Bone defect along the sagittal and lambdoid sutures in a 2-year-old girl with neurofibromatosis type 1. Anteroposterior skull radiograph shows multiple bone defects along the sagittal and lambdoid sutures (arrows). Mild dysplasia of the right sphenoid wing is also noted.
Sphenoid wing dysplasia is almost always associated with plexiform neurofibromas of the orbit and periorbital region (see Fig. 39-5). Extension of plexiform neurofibromas to the cavernous sinus and the third to sixth cranial nerves is often observed. On CT, they are of low attenuation and do not show enhancement after administration of intravenous contrast material.
MRI
Plexiform neurofibromas present as heterogeneous masses, displaying mostly a low signal intensity compared with that of the brain parenchyma on T1W images and a high signal intensity on T2W images. However, the signal intensities are variable. T2W images typically show a peripheral high signal and a central low signal intensity compared with that of the muscle, called “target sign” (see Fig. 39-5B, C). The central hypointensity may reflect a dense central core of collagen.4 After administration of intravenous contrast material, contrast enhancement of the tumor is variable but usually some parts of the tumor are enhanced (see Fig. 39-5D).
Vascular Dysplasia
CT
NF1 patients occasionally show dysplasia of the cerebral vasculature. The dysplasia is characterized by intimal proliferation resulting in arterial stenosis. The common and internal carotid arteries and the proximal middle and anterior cerebral arteries are usually involved (Fig. 39-E6).
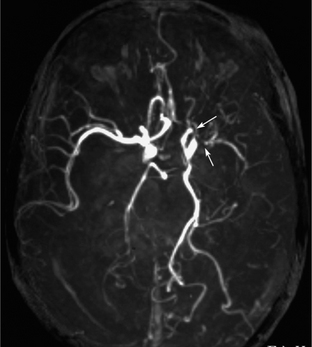
FIGURE 39-E6 Vascular dysplasia of neurofibromatosis type 1. MR angiography demonstrates stenosis of the distal portion of the left internal carotid and proximal portion of the left middle cerebral arteries (arrows).
Spinal Manifestations
Nontumoral spinal manifestations in NF1 patients include scoliosis, enlargement of the neural foramina (with or without lateral meningoceles), and posterior scalloping of the vertebral bodies. Both nerve sheath tumors and even spinal cord tumors may develop in NF1 patients but usually become symptomatic in older age.5
MRI
If an intramedullary tumor is found in a patient with NF1, it is likely to be an astrocytoma (commonly of low grade). The imaging findings of intramedullary astrocytoma in NF1 patients are identical to those in patients without NF1. If syringohydromyelia is found without an intraspinal mass in patients with NF1, a thin-slice contrast-enhancement MRI should be obtained to search for underlying intramedullary lesions.4
Nerve sheath tumors such as neurofibromas, plexiform neurofibromas, and MPNSTs may be found. Spinal/paravertebral neurofibromas are almost characteristic of NF1 patients. They may be single or multiple and unilateral or bilateral, and they may involve both intraspinal and extraspinal regions (dumbbell tumor) (Fig. 39-6). When small, the neurofibromas present as nodules along the nerve roots of the cauda equina on MRI. When large, they involve the neural foramina and extend into the paravertebral region.4 As a result, the neural foramina are enlarged by bone erosion. On MRI, neurofibromas show a slight hyperintensity compared with the muscle on T1W images and a peripheral hyperintensity with a relatively hypointense center on T2W images (see Fig. 39-6). This finding on T2W images is called a “target sign.” Cord compression may occur if bilateral tumors grow at the same level.
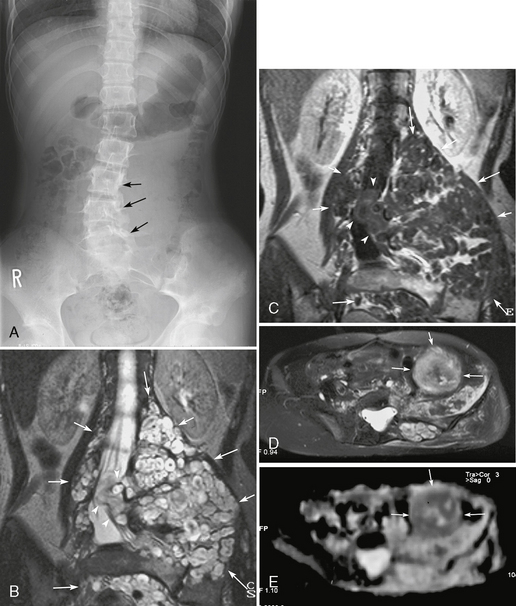
FIGURE 39-6 Neurofibromatosis type 1. Lumbar scoliosis, multiple paravertebral neurofibromas, and development of a malignant peripheral nerve sheath tumor are shown on a radiograph of the lumbar spine (A), T2W (B) and gadolinium-enhanced T1W (C) coronal MR images, and follow-up axial T2W MR image (D) and apparent diffusion coefficient map (E) 3 years later. Numerous plexiform neurofibromas showing target signs are noted in the bilateral paravertebral regions and pelvis (arrows, B, C). Some tumors extend to the spinal canal (arrowheads, B, C). The lumbar spine shows secondary scoliosis with bone erosion (arrows, A). A follow-up MRI examination after 3 years shows an enlarged tumor with heterogeneous T2 prolongation and low apparent diffusion coefficient (arrows, D, E). The tumor was resected and proved to be a malignant peripheral nerve sheath tumor.
MPNSTs occur in NF1 patients with a reported incidence of 10% to 15%.4 Compared with benign neurofibromas, MPNSTs tend to be larger and relatively well circumscribed (see Fig. 39-6D, E). Increased internal heterogeneity has been reported to warrant a diagnosis of MPNST, although the finding is present in rare benign neurofibromas. The target sign is less common (see Fig. 39-6D, E). Local pain and rapid growth are most suggestive of malignant degeneration.4
NEUROFIBROMATOSIS TYPE 2
Epidemiology
A recent update suggests that the incidence may be as high as 1 : 25,000, and at least higher than 1 : 100,000.12
Clinical Presentation
The most sensitive modified clinical diagnostic criteria for NF2 are as follows13,14:
• Bilateral vestibular schwannomas
• A first-degree relative with NF2 and
• Unilateral vestibular schwannoma and any two of: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities*
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


