CHAPTER 24 Vascular Malformations
Four major types of primary malformations of the vascular systems can be identified: arterial, arteriovenous, capillary, and venous. Each of these malformations manifests intrinsic primary abnormalities and each causes derivative secondary change in the rest of the vascular system. Classically, these vascular malformations are grouped into four categories (1) the dural and pial arteriovenous malformations and shunts, (2) cavernous malformations (cavernomas), (3) capillary telangiectasias, and (4) developmental venous anomalies.
ARTERIOVENOUS MALFORMATIONS AND FISTULAS
Types
Pial Malformations
AVMs are characterized by a network of abnormal channels (the nidus) interposed between the arterial feeder(s) and the draining vein(s). The shunts may be small (micro-AVMs) with a nidus less than 1 cm in diameter and normal size arteries and draining veins, or the shunts may be larger (macro-AVMs) with nidi larger than 1 cm in diameter and enlarged feeding arteries and draining veins. AVMs may be compartmentalized into portions fed and drained by separate vessels, as documented by angiography or at surgery. In many cases of pial AVMs the separate compartments exhibit differing internal angioarchitecture with more fistulous or more glomerular nidal appearances (see later) within the different compartments (Fig. 24-1).
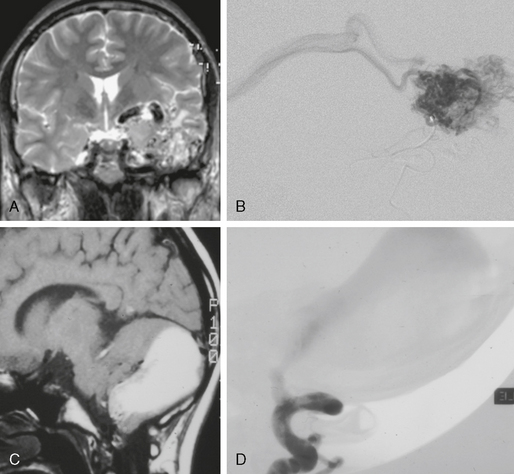
FIGURE 24-1 Glomerular versus fistulous arteriovenous malformation. Whereas on MRI glomerular AVMs can be perceived as a tangle of abnormal vessels within the brain parenchyma (A, B), fistulous AVMs are typically recognized by their large venous pouch that may demonstrate a mixed signal depending on the flow properties within the pouch (C, D). The superselective injection demonstrates the nidus aspect in the glomerular AVM whereas in the fistulous AVMs the direct transition from the artery into the massively enlarged venous pouch can be visualized.
AVFs consist of direct, fistulous transitions from an artery into a vein. They, too, can be classified as micro-AVF or macro-AVF depending on the size of the feeding artery. The AVFs are found almost exclusively on the surface of the brain or the spinal cord. Pial AVFs appear to be relatively frequent in children and rare in adults (Fig. 24-2).
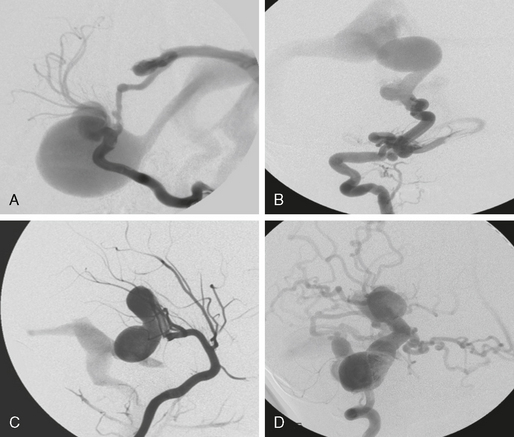
FIGURE 24-2 Four different fistulous AVMs. These types of AVMs are, owing to their clinical symptomatology, most often encountered in the pediatric age group. When multiple fistulas occur, the possible diagnosis of Osler’s disease (hereditary hemorrhagic telangiectasia) has to be discussed. Most often, these fistulas are single-hole fistulas (i.e., multiple feeding arteries converging at one single spot into the enlarged venous pouch). Clinical manifestations are due to venous congestion that may lead to hydrodynamic disorders in the pediatric population. Spontaneous thrombosis may also cause life-threatening mass effect.
Arteriovenous Malformations that Involve the Cortex
Cortical arteriovenous lesions involve the cortex, are fed exclusively by cortical arteries, and drain into superficial veins (unless secondary thrombosis of cortical veins shunts the venous flow into alternate pathways). These lesions are also designated sulcal AVMs (Fig. 23-3).
Cortical-subcortical arteriovenous lesions recruit cortical arteries and drain into superficial veins but may also drain into the deep venous system if the transcerebral venous system is patent. These lesions are also designated gyral AVMs (Fig. 23-4). In both cortical (sulcal) and cortical-subcortical (gyral) lesions, some regions of the cortex drain to deeply located veins that should not be considered as true parts of the deep venous system. Such vessels include the medial veins of the temporal lobe and the basal vein of Rosenthal, the veins of the cerebellar vermis, and the precentral cerebellar veins.
Corticoventricular arteriovenous lesions correspond to the classic pyramidal malformations that are based on the cortex and reach the ventricular wall at their apex. These malformations are fed by both cortical and perforating arteries and drain to both superficial and deep veins. Corticocallosal AVMs belong to the corticoventricular group (Fig. 23-5). They have the same venous characteristics but do not recruit perforating arteries. Corticocallosal AVMs drain into the subependymal veins and later into the deep venous system. The arterial supply to the corpus callosum is linked to the cortical arterial network, even though it may simulate perforating arterial channels in the supraoptic region and choroidal arteries at the splenium.
Deep-Seated Arteriovenous Lesions
Deep-seated lesions are located in the depth of the telencephalon, diencephalon, brain stem, or cerebellum (Fig. 23-6). The nidi involve the deep nuclei, and the arterial and venous connections are along the long fiber tracts. The deeply seated arteriovenous lesions recruit perforating arteries exclusively and drain into the deep venous system. They may use transcerebral veins, if patent, as direct venous outlets or as collateral pathways. Arteriovenous lesions of the lenticular and striate nuclei may recruit transcortical arteries from the insular branches of the middle cerebral artery. Arteriovenous lesions of the dentate nucleus may recruit hemispheric collaterals of the cerebellar arteries. MRI and angiography will be able to distinguish these deep-seated lesions from cortical-subcortical lesions, particularly because the dominant supply of the deep intracerebral lesions arises from the perforators.
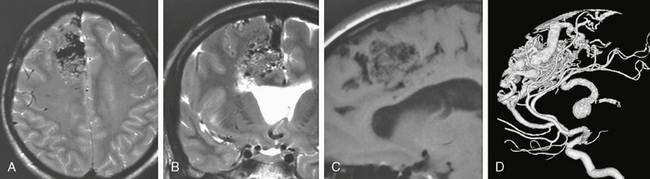
FIGURE 24-4 A to D, This corticocallosal AVM had a massively dilated thalamostriate vein draining into the internal cerebral vein that subsequently led to unilateral hydrocephalus. On MRI, the flow voids can be well appreciated on T2W images (A, B) while on T1W nonenhanced studies (C) the AVM signal is typically hypointense.
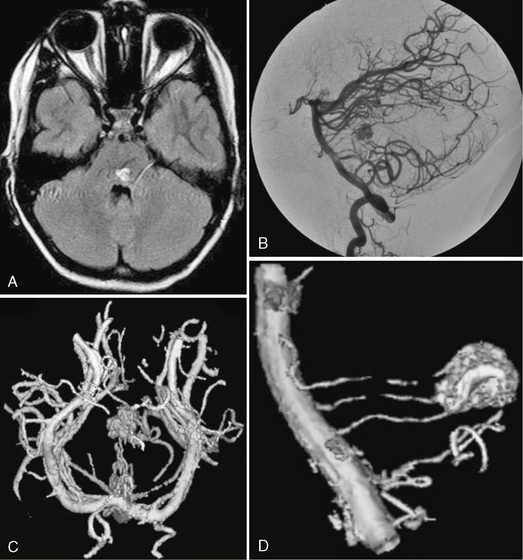
Arteriovenous Lesions of the Choroid Plexus
Choroid plexus AVMs are important to recognize because they are anatomically extracerebral lesions accessible to surgical and endovascular therapy. Choroidal arteriovenous lesions are fed primarily by choroidal arteries and by subependymal arterial feeders arising from the circle of Willis, including the anterior thalamoperforate arteries arising from the posterior communicating arteries that course through the walls of the third ventricle to reach its roof and the adjacent choroid fissure (Fig. 23-7). By definition, choroid plexus arteriovenous lesions have no cortical arterial supply. They drain via ventricular veins, with occasional recruitment of transcerebral veins. These extra-axial lesions can be distinguished from intra-axial AVMs by angiographic criteria.
Pathophysiology
The vessels surrounding the AVM are affected by the congenital malformation and by the hemodynamic alterations it produces. These secondary, acquired effects influence the clinical presentation and outcome. Pial AVMs may be found incidentally or because of headache, seizures, or hemorrhage. Children, especially those with high-flow fistulas, may manifest somatic symptoms and psychomotor retardation when venous congestion is a prominent feature of the malformation. Headaches are more commonly seen in children and are typically pseudomigrainous. AVMs in occipital locations and AVMs that have transdural supply, venous ectasia, and venous congestion show increased incidence of headaches. Most seizures associated with brain AVMs are focal and may reflect venous congestion or small hemorrhages that irritate healthy brain tissue near the brain AVM.
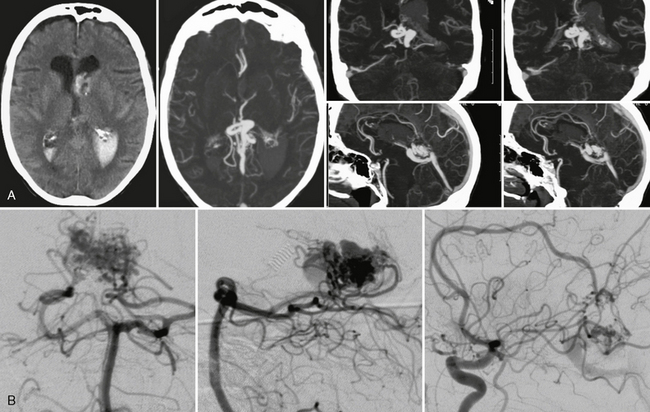
FIGURE 24-7 Acute intraventricular hemorrhage in a 68-year-old woman without prior history of hypertension led to CTA that was able to demonstrate a choroidal type AVM with large venous outpouchings (A). CTA demonstrates a tangle of pathologic and enlarged vessels as a sign for the AVM that was verified on DSA (B). Typical for a choroidal AVM, feeders are recruited from the pericallosal artery via the limbic arch and from the lateral posterior choroidal arteries of the posterior cerebral artery.
Classification
The Spetzler-Martin classification of pial brain AVMs is based on the size of the AVM, the pattern of venous drainage, and the eloquence of the portions of brain adjacent to the AVM. The classification was devised to anticipate the risk of treating brain AVMs surgically but has been extended to predict the (presumed) natural history of the entire group of AVMs. We believe this generalization to be improper because (1) the classification does not assess the special characteristics of an AVM in an individual patient (e.g., associated aneurysms), (2) the classification does not recognize that an AVM with high-grade risk for surgery is not necessarily dangerous to the patient, and (3) the classification does not allow assessment for alternate therapy by endovascular techniques and radiosurgery.
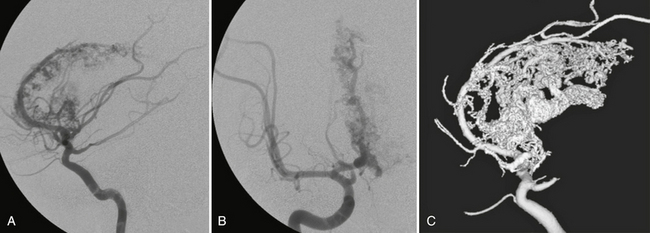
To determine whether endovascular therapies are suitable for a brain AVM, one must assess the angioarchitecture of the malformation, including the nature of the feeding artery, the number of separate compartments of the malformation, any arterial or venous ectasias near to or within the malformation, and the nature of the venous drainage. There are two basic types of feeding artery. Direct arterial feeders end in the AVM. Indirect arterial feeders supply the normal cortex and also supply the AVM “en passage” via small vessels that arise from the normal artery (see Fig. 24-5). Intranidal arterial aneurysms or venous varices indicate weak points in the system. Drainage into the deep venous system and stenoses that restrict venous outflow indicate increased risk of spontaneous hemorrhage. A long pial course of the draining vein may indicate that venous drainage is restricted over a large area, increasing the risk of venous congestion and subsequent epilepsy. Conversely, a short vein that drains almost directly into a dural sinus is unlikely to interfere with the normal pial drainage.
Imaging
CT
Small AVMs may be difficult to discern (Fig. 24-8). Larger AVMs usually display (1) tangled “serpiginous” parenchymal vessels that appear slightly dense due to blood pooling and (2) large draining veins in the subarachnoid space. Calcifications are present in about one third of brain AVMs. Focal hemorrhage, focal mass, or focal atrophy may also be present. Contrast enhancement opacifies the enlarged feeding arteries, the tangle of vessels in the parenchyma, and the dilated draining veins.
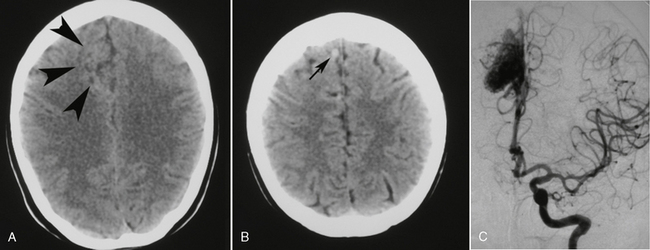
FIGURE 24-8 This patient presented to the emergency department with a first-ever seizure. A and B, Routine non–contrast-enhanced cranial CT revealed an ill-defined area of gray matter/white matter differentiation in the right frontal lobe (arrowheads) and a tubular extra-axial structure (small arrow) extending to the superior sagittal sinus, indicative of a vascular malformation with a large draining vein. This was verified on DSA (C).
MRI
T2-weighted (T2W) imaging shows punctate to linear flow voids in the subarachnoid space over the brain, reflecting the dilated surface vessels. Within the brain, the punctate flow voids often lie within hyperintense, gliotic parenchyma. This gliosis may extend to the surrounding brain tissue. T1-weighted (T1W) imaging shows highly variable signal within the AVM, due to turbulent flow, blood degradation products, and the variable flow rate in the veins (Fig. 24-9). There is usually intense contrast enhancement of the vessels themselves. T2*-weighted images may detect smaller areas of hemorrhage.
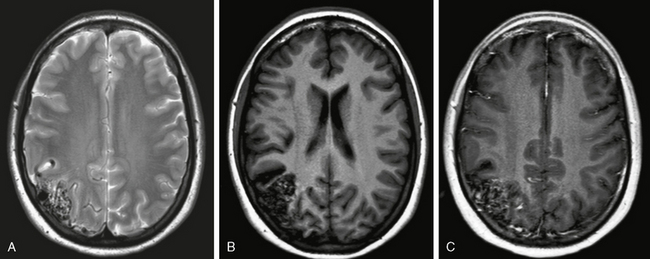
FIGURE 24-9 A to C, MR aspect of a cortical AVM. A conglomeration of dilated vascular masses can be seen on both T2W (A) and T1W (B, C) images. The signal (especially on T1W sequences) depends on the velocity and direction of flow with both flow-void and/or enhanced areas. Remnants of recent or old bleeding have to be specifically sought using T2W GRE sequences (not shown) because AVMs that have bled once have a higher propensity to rebleed. FLAIR and T2W sequences should be evaluated for perifocal gliosis. Classically, the cortical AVMs extend wedge shaped into the subcortical areas within a sulcus.
MRA
“Static” magnetic resonance angiographic (MRA) sequences (such as time-of-flight [TOF] and phase contrast [PC] MRA) may detect the lesion but do not detail the angioarchitecture of the malformation (see Fig. 24-3). Static MRA provides little information about the presence of flow-related or intranidal aneurysms or the direct/indirect nature of the feeding arteries. Venous stenoses and ectasias are poorly displayed due to turbulent flow. Static MRA also fails to define the hemodynamics of the AVM, including the principal feeding arteries, the early venous drainage pattern, and the flow velocity.
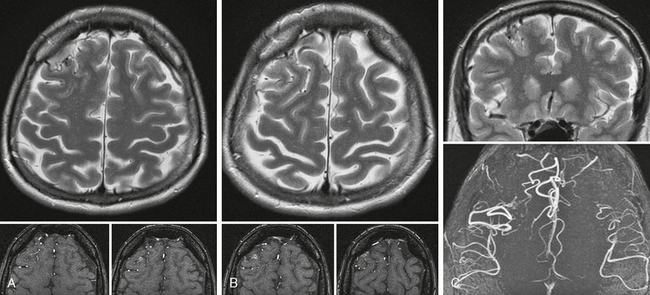
FIGURE 24-3 A to C, Cortical micro-AVM. On T2W images the cortical location of this small AVM can be best appreciated on the coronal cuts; however, the axial slices also demonstrate the abnormal tangle of vessels. The axial raw data of the TOF sequence and their reconstruction demonstrate the asymmetric number of flow voids and the abnormal cortical vessels in the right frontal lobe, suggesting the diagnosis.
High-resolution, 3D, dynamic contrast-enhanced MRA provides greater information about the velocity and direction of blood flow through each component of the AVM and can confirm the diagnosis of an AVM suggested by prior conventional MRI and MRA (Fig. 24-10). Complex subtraction techniques help to eliminate artifactual signal resulting from intraparenchymal hemorrhage. However, caution must be taken in very fast-flow AVMs, because the rapid flow results in poorer contrast of the feeding arteries compared with normal vessels and draining veins (Fig. 24-11).
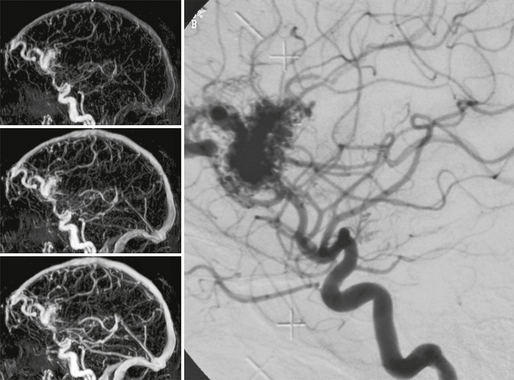
FIGURE 24-10 Contrast-enhanced dynamic MRA can depict the large feeding vessels, the size of the nidus, its location, and the major draining veins. Pure morphologic classifications are therefore possible using this technique.
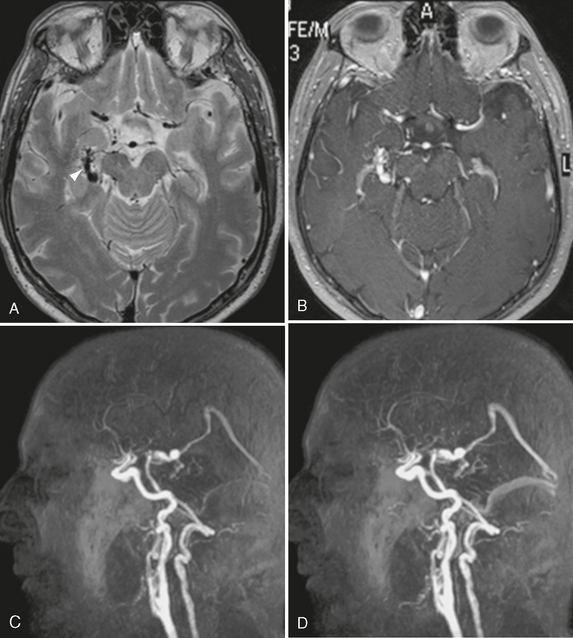
FIGURE 24-11 The value of MR contrast-enhanced angiography is shown in this case. Whereas classic MR sequences such as T2W and T1W images (A, B) are able to show only a dilated basal vein of Rosenthal (arrowhead, A), the dynamic aspect of the contrast-enhanced MRA (C, D) is clearly able to depict the early arteriovenous shunt into the basal vein of Rosenthal and the venous filling of the straight and transverse sinuses in the early arterial phase, which is suggestive of a large shunting volume.
Functional imaging methods such as blood oxygenation level–dependent contrast (BOLD) fMRI and perfusion imaging may give additional information about the pathophysiology of these AVMs (Fig. 24-12).
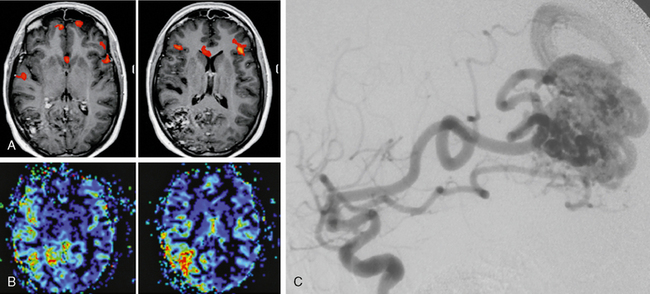
FIGURE 24-12 A to C, This 27-year-old woman underwent MRI due to recurrent and worsening headaches and repeated episodes of speech arrest followed by confusion. MRI demonstrated a right parietal AVM that, owing to its location, was unable to explain the clinical symptoms of recurrent speech arrests in this right-handed patient. Functional MRI was performed to localize the speech-related areas and demonstrated bilateral representation of areas responsible for naming located in the pars triangularis of the inferior frontal gyrus (Broca’s area and its contralateral homologue). A perfusion MRI demonstrated an increased flow not only within the AVM but also in its vicinity, namely, those areas that were activated during the speech tasks. Owing to these results it was hypothesized that the high shunting volume of the AVM led to a reduced capillary transit time in the vicinity of the AVM and therefore to the recurrent speech arrests. After embolization achieved a significant reduction of the shunting volume the patient was free of symptoms. The remainder of the AVM was treated with gamma knife radiosurgery.
Conventional Digital Subtraction Angiography
Conventional catheter angiography remains the study of choice to evaluate all potential feeding arteries, including external carotid branches that can supply the AVM via dural and leptomeningeal collaterals. Digital subtraction angiography (DSA) also provides a full assessment of the other vascular territories to rule out multiple shunts that may indicate an underlying systemic disease (e.g., hereditary hemorrhagic telangiectasia) or a syndromic disease such as cerebrofacial arteriovenous metameric syndrome. DSA should define the angioarchitecture of all feeding arteries, determine whether the shunt is a direct or indirect type, and display all significant arterial dilatations, stenoses, and associated flow-related aneurysms (Fig. 24-13). DSA should define the nidus, including its size, fistulous versus glomerular nature, intranidal aneurysms, false aneurysms (in case of hemorrhagic AVMs), associated angioectasia, and neoangiogenesis. DSA should also display all of the draining veins to resolve deep versus superficial patterns of drainage, length of the pial segments, venous ectasias, and any stenoses that might cause flow restriction. DSA is paramount, therefore, for ascertaining the risk posed by an individual brain AVM and for selecting the therapy best suited for that specific AVM (Fig. 24-14).
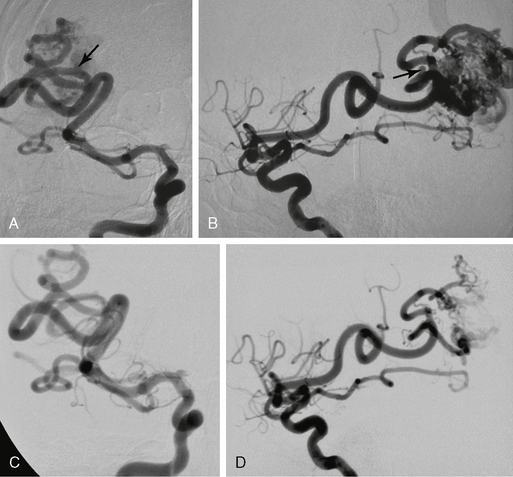
FIGURE 24-13 Effects of embolization on the remodeling of the feeding vessels. In this AVM an aneurysm can be seen in each division of the two major feeding arteries (arrows in A and B). Six weeks after initial embolization with significant flow reduction, the aneurysms are no longer seen, which is indicative of its flow-related nature (C, D).
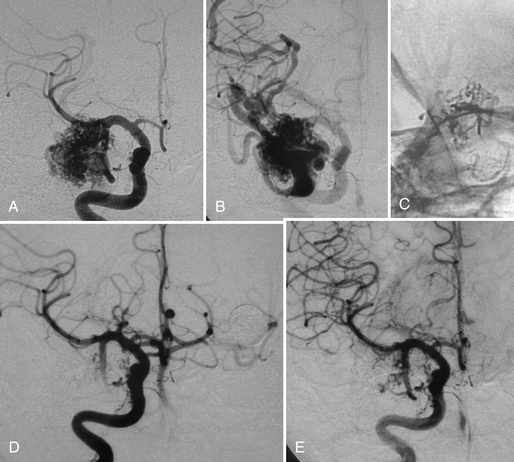
FIGURE 24-14 Right temporal AVM before (A, B) and after treatment (C to E) with superselective glue injection demonstrating a markedly reduced size of the nidus and an enhanced flow in other cortical areas immediately after the intervention.
Two conditions merit special imaging consideration:
1. The acute hemorrhage that obscures an AVM. A small AVM may be completely obscured by the mass effect of an acute hematoma. In the setting of acute hemorrhage, therefore, the initial CT, MR, MRA, and DSA may be insufficient to rule out an AVM, even if contrast enhancement is used. For these reasons, a second DSA should be performed after the clot has cleared, especially if the patient is a child or the clot was atypical in appearance.
2. Post-treatment evaluation. Patients presenting with a treated AVM are evaluated for the completeness of occlusion and restoration of flow previously diverted toward the shunt (steal phenomenon). In these cases, dynamic contrast-enhanced MRA may be sufficient to separate the arterial and venous phases and document the presence of any persistent early filling of the draining veins. If the MRA shows an early vein, the AVM is not completely occluded. If dynamic contrast-enhanced MRA fails to demonstrate an early-filling vein, it may still be necessary to use the superior spatial resolution of DSA to ensure that the shunt is completely obliterated (Fig. 24-15).
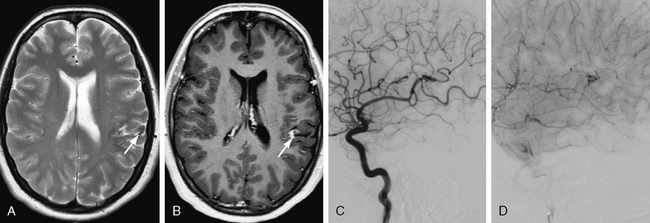
FIGURE 24-15 Effects of gamma knife surgery. Three years before this MRI, stereotactic radiation of a left parietal AVM was performed. In the control image, a hemosiderin stain and persistent contrast enhancement in the area of the former AVM can be seen (arrows in A and B). DSA confirmed the complete occlusion of the AVM (C, D), but a slight area of hyperperfusion resembling the contrast enhancement on MRI was seen. Because no early-draining vein could be visualized, the treatment was, however, considered completed.
Differential Diagnosis
Cerebral Proliferative Angiopathy
Cerebral proliferative angiopathy (CPA, diffuse nidus type AVM) is a clinical entity distinct from the “classic” brain AVMs in its clinical presentation, natural history, angioarchitecture, and treatment. CPA is present in 2% to 4% of all brain AVMs. It is more common in females by 2 : 1 and presents at a mean age of 20 years. Seizures, headaches, and transient ischemic attacks are far more frequent in CPA than in classic AVMs, whereas hemorrhages are exceedingly rare. On CT and MRI, CPA presents as a diffuse network of densely enhancing vascular spaces intermingled with normal brain parenchyma. T1W and T2W images show small, widely distributed flow voids that may involve multiple lobes or the entire hemisphere (Fig. 24-16). In most cases, the primary lesion extends from the surface into the basal ganglia and thalamus and involves more than one vascular territory. Compared with the size of the nidus, relatively few draining vessels are seen on CT or MRI, and these are only moderately enlarged. Perfusion-weighted MRI demonstrates perfusion abnormalities far beyond the boundaries of the morphologic malformation seen on conventional MR sequences, so the disease actually affects the entire hemisphere. The nidus shows increased cerebral blood volume, slightly decreased time to peak (TTP), and prolonged mean transit time (MTT). Remote from the nidus, in normal-appearing cortical and subcortical areas, the TTP is increased and the blood volume is decreased, indicating remote, widespread hypoperfusion (Fig. 24-17). Angiography shows no dominant feeders. Instead, CPA is fed by multiple arteries that are not enlarged or are only moderately enlarged. Unlike classic AVMs, all arteries of the affected region contribute equally to the malformation. Stenoses of the proximal arteries are present in 40% and affect the internal carotid artery (ICA) and the proximal horizontal segments of the middle cerebral artery (M1) and anterior cerebral artery (A1). Most cases show transdural supply to both the malformation and the normal brain tissue. The nidus has a classic appearance with scattered areas of “puddling” of contrast medium within what looks like capillary ectasias. These persist into the late arterial and early venous phases. The nidus is usually fuzzy, poorly circumscribed, and larger than 6 cm in diameter. Intranidal vessels show a capillary angioectasia. Perinidal angiogenesis is often present and difficult to distinguish from the nidus proper. There is no high-flow fistulous component to the arteriovenous shunt, so early opacification of draining veins is uncommon. The size of the draining veins, the “shunt volume,” and the time until the veins are visualized never “correspond” to the size of the nidus. Histopathology demonstrates the presence of normal-appearing neural tissue intermingled between these vascular channels. Perivascular gliosis is only mild. There is additional capillary angiogenesis within the subcortical region (Fig. 24-18). The implication is that the brain tissue within the “nidus” of the CPA is functional, similar to the brain tissue found between the abnormal vascular channels of capillary telangiectasia. This alters therapeutic strategies. Because these lesions appear to result from ischemia, therapy should be directed toward revascularization procedures, such as burr-hole therapy, not at the lesion proper.
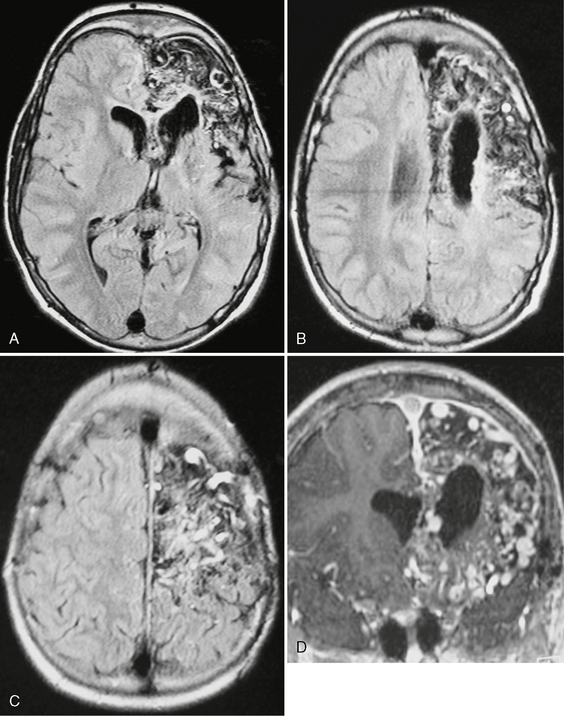
FIGURE 24-16 FLAIR-weighted images (A–C) and T1W MR image post contrast (D) in a 15-year-old boy with recurrent seizures, disabling headaches, and transitory ischemic attacks. A diffuse network of densely enhancing vascular spaces can be seen throughout the frontal lobe. Normal brain tissue seems to be dispersed along the vascular malformation. The size of the malformation is atypical for an AVM. These findings correspond with the diagnosis of cerebral proliferative angiopathy.
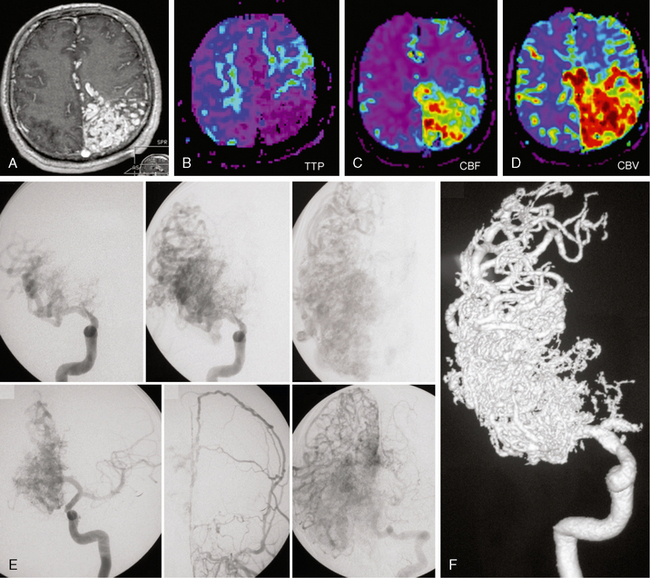
FIGURE 24-17 Cerebral proliferative angiopathy. T1W (A) and perfusion MRI with time to peak (TTP), cerebral blood volume (CBV), and cerebral blood flow (CBF) maps in a 11-year-old girl with headaches demonstrating a large left frontoparietal nidus with brain parenchyma intermingled between the vascular spaces. Perfusion-weighted MRI demonstrates an increase in cerebral blood volume (D) and flow (C), with hypervascularization within the nidus and decreased times to peak (B) in the area surrounding the nidus, indicating the ischemic nature of the disease. E, F, Angiography in the early arterial phase demonstrates the absence of dominant feeders and the equal contribution of many different arteries. The contrast dynamics reveal persistence of contrast material in the malformation and no early venous drainage. Transdural supply testifies for the proliferative component of the disease while injection into the vertebral artery demonstrates diffuse neoangiogenesis in other cortical areas.
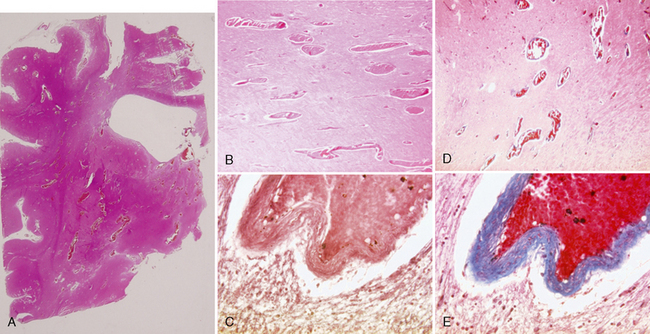
FIGURE 24-18 Histologic specimens of cerebral proliferative angiopathy. Paraffin-embedded tissues shown here were stained with hematoxylin and eosin (A, B), orcein van Gieson (C), and Masson trichrome (D, E). In A, a coronal cut through the right hemisphere at the level of the frontal horn and the striatum demonstrates intraparenchymal vascular proliferation with dilated and irregular arterioles that are otherwise histologically normal. These dilated vascular channels are located preferentially in regions with low cellular density (white matter tracts and between the basal ganglia). The venous vessel walls demonstrate collagenous thickening of the veins. Mild perifocal gliosis is seen directly surrounding the vessels, but normal-appearing neuronal tissue can be seen intermingled between the vascular channels.
The combination of segmental stenoses of proximal arteries and distal angioectasia suggests that the pathogenesis of this lesion may be abnormal proliferation of the vessel wall. In their 1975 classification of vascular lesions in childhood and infancy, Mulliken and Glowacki differentiated between (1) vascular lesions that demonstrate cellular proliferation and endothelial hyperplasia (“hemangiomas” or true vascular tumors as in PHACES syndrome) and (2) vascular lesions that have normal endothelial turnover but structural abnormalities of the capillary, venous, lymphatic, or arterial channels (e.g., vascular malformations like brain AVMs). CPA can be regarded as an intermediate or transitional form between these two major types of vascular lesion. CPA exhibits both proliferative features (less prominent than in hemangiomas) and malformative features (less prominent than in true AVMs).